










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkInsuficiencia cardíaca
versión On-line ISSN 1852-3862
Insuf. card. vol.11 no.4 Ciudad Autónoma de Buenos Aires dic. 2016
ARTICULO DE ACTUALIZACION
Disfunción mitocondrial y enfermedades cardiovasculares
Alfredo D'Ortencio1, Alfredo Navigante2
1 Médico cardiólogo. Jefe del Servicio de Cardiología. Instituto de Oncología "Ángel H. Roffo". Facultad de Medicina. Universidad de Buenos Aires (UBA). Ciudad de Buenos Aires. República Argentina.
Director de la Unidad Docente Hospitalaria. Facultad de Medicina. Universidad de Buenos Aires (UBA). Ciudad de Buenos Aires. República Argentina.
Director General del Instituto de Oncología "Ángel H. Roffo". Facultad de Medicina. Universidad de Buenos Aires (UBA). Ciudad de Buenos Aires. República Argentina.
2 Médico cardiólogo. Unidad de Investigación de Transferencia (UIT). Instituto de Oncología "Ángel H. Roffo". Facultad de Medicina. Universidad de Buenos Aires (UBA). Ciudad de Buenos Aires. República Argentina.
Correspondencia: Dr. Alfredo D'Ortencio.
Instituto de Oncología "Ángel H. Roffo".
Av. San Martín 5481. CP: 1417. Ciudad de Buenos Aires. República Argentina.
E-mail: adortencio@intramed.net
Recibido: 09/02/2016
Aceptado: 20/06/2016
Resumen
Las mitocondrias son organelas celulares encargadas de suministrar la mayor parte de la energía necesaria para la actividad celular (respiración celular), actuando como centrales energéticas de la célula y sintetizando adenosín trifosfato (ATP) a expensas de los carburantes metabólicos. Estas organelas móviles existen en un sistema de interconexión dinámica dentro de cada célula, participando continuamente de uniones por procesos de fusión y divisiones por procesos de fisión. Estos eventos participan en una amplia variedad de procesos biológicos, que incluyen el desarrollo embrionario, el metabolismo, la apoptosis y la mitofagia. La disfunción mitocondrial es considerada una vía común en una serie de enfermedades, tanto agudas como crónicas, por el fenómeno del estrés oxidativo y la generación de la respuesta inflamatoria sistémica. Esta disfunción está involucrada en la patogenia de diferentes enfermedades y condiciones genéticas como las enfermedades neurodegenerativas, neoplásicas, endocrinas y cardiovasculares, entre otras. El desarrollo de tratamientos metabólicos y farmacológicos eficaces para enfermedades mitocondriales definidas es una promesa para futuras terapias que podrían mejorar los síntomas de una amplia gama de alteraciones metabólicas y trastornos degenerativos e incluso el envejecimiento. En esta actualización se revisa cómo la dinámica mitocondrial se altera en diversas enfermedades cardíacas y cómo este conocimiento podría proporcionar nuevos objetivos terapéuticos para su tratamiento.
Palabras clave: Mitocondria; Dinámica mitocondrial; Disfunción mitocondrial; Inflamación; Hipertensión pulmonar; Ductus arterioso; Diabetes; Insuficiencia cardíaca; Cáncer
Summary
Mitochondrial dysfunction and cardiovascular disease
Mitochondria are cellular organelles in charge of providing the greater part of the energy required for cellular activity (cellular respiration), acting as power stations of the cell by synthesizing adenosine triphosphate (ATP) at the expense of metabolic fuels. These mobile organelles exist in dynamic networks. They continuously join by the process of fusion and divide by the process of fission. These events are involved in a wide variety of biological processes, including embryonic development, metabolism, apoptosis and mitophagy. Mitochondrial dysfunction is considered esa common pathway in a number of diseases, both acute and chronic, by the phenomenon of oxidative stress and the generation of systemic inflammatory response. This dysfunction is involved in the pathogenesis of different diseases and genetic conditions such as neurodegenerative, neoplastic, endocrine and cardiovascular diseases, among others. The development of effective metabolic and pharmacological treatments for defined mitochondrial diseases holds promise for future therapies that could improve the symptoms of a broad range of metabolic and degenerative disorders, and even aging. This update revises how altered mitochondrial dynamics in several heart diseases and how this knowledge could provide new therapeutic targets for treatment.
Keywords: Mitochondria; Mitochondrial dynamics; Mitochondrial dysfunction; Inflammation; Pulmonary hypertension; Ductus arteriosus; Diabetes; Heart failure; Cancer
Resumo
Disfunção mitocondrial e doença cardiovascular
As mitocôndrias são organelas celulares responsáveis de fornecer a maior parte da energia necessária para a atividade celular (respiração celular), atuando como usinas de energia celular e sintetizando adenosina-trifosfato (ATP), a expensas de combustíveis metabólicos. Essas organelas móveis existem num sistema de interconexão dinâmica dentro de cada célula, participando de forma contínua de uniões pelo processo de fusão e divisões pelo processo de fissão. Estes eventos são envolvidos em uma ampla variedade de processos biológicos, incluindo o desenvolvimento embrionário, o metabolismo, a apoptose e mitofagia. A disfunção mitocondrial é considerada uma via comum num certo número de doenças, tanto aguda como crônica, pelo fenômeno do stress oxidativo e a geração de resposta inflamatória sistêmica. Esta disfunção está envolvida na patogênese de várias doenças e condições genéticas tais como doenças neurodegenerativas, neoplásicas, endócrinas e cardiovasculares, entre outras. O desenvolvimento de tratamentos metabólicos e medicamentosos eficazes para doenças mitocondriais definidas é uma promessa para futuras terapias que podem melhorar os sintomas de uma ampla gama de alterações metabólicas e distúrbios degenerativos e até mesmo envelhecimento. Esta atualização revisa a alteração da dinâmica mitocondrial em diversas doenças cardíacas e como esse conhecimento poderia fornecer novos alvos terapêuticos para seu tratamento.
Palavras-chave: Mitocôndria; Dinâmica mitocondrial; Disfunção mitocondrial; Inflamação; Hipertensão pulmonar; Ducto arterial; Diabetes; Insuficiência cardíaca; Câncer
Introducción
La disfunción mitocondrial es considerada una vía común en el fenómeno de estrés oxidativo y la generación de la respuesta inflamatoria sistémica; vía común a una serie de entidades, tanto agudas como crónicas, teniendo como paradigma desde sepsis y leucemias agudas hasta neoplasias sólidas e insuficiencia cardíaca1,2.
Esta disfunción mitocondrial, tanto en la función como en la estructura de las mitocondrias, se ha encontrado cada vez con mayor frecuencia asociadas a enfermedades cardiovasculares. Algunas anomalías mitocondriales pueden tener una base genética, que producen una disfunción de la fosforilación oxidativa o de los defectos en la oxidación de los ácidos grasos debidos a mutaciones específicas del ADN nuclear, mientras que otras de estas anomalías parecen ser debidas a causas cardiotóxicas, ambientales o desconocidas3.
El corazón depende estrechamente de la energía oxidativa generada en las mitocondrias, principalmente a partir de la beta oxidación de los ácidos grasos, de la cadena respiratoria de electrones y de la fosforilación oxidativa. En esta actualización, se revisa cómo la dinámica mitocondrial se altera en diversas enfermedades cardíacas y cómo este conocimiento podría proporcionar nuevos objetivos terapéuticos para su tratamiento.
Mitocondria
Las mitocondrias son organelas celulares encargadas de suministrar la mayor parte de la energía necesaria para la actividad celular (respiración celular). Actúan, por lo tanto, como centrales energéticas de la célula y sintetizan adenosín trifosfato (ATP) a expensas de los carburantes metabólicos (glucosa, ácidos grasos y aminoácidos). La mitocondria presenta una membrana exterior permeable a iones, metabolitos y muchos polipéptidos (Figura 1). Eso es debido a que contiene proteínas que forman poros llamados porinas o VDAC (voltage-dependent anion channels: canal aniónico dependiente de voltaje), que permiten el paso de moléculas de hasta 10 kDa de masa y un diámetro aproximado de 2 nm4.

Figura 1. Célula eucariota con sus elementos fundamentales: membrana plasmática, citoplasma y núcleo. Dentro del citoplasma o citosol encontramos a las mitocondrias. Éstas se presentan de forma ovoide, de bastón o esféricas, rodeadas por 2 membranas: una externa (lisa) y una interna (con pliegues llamados crestas mitocondriales). Por dentro de esta última se encuentra la matriz mitocondrial. Sobre la membrana interna y en la matriz se ubican una gran cantidad de enzimas que participan en la llamada: respiración celular. La mitocondria es la principal proveedora de ATP de la célula. Poseen su propio ADN, así como ribosomas y otros tipos de ARN.
Estas organelas móviles existen en un sistema de interconexión dinámica dentro de cada célula, participando continuamente de uniones por procesos de fusión (sinónimos: unión, integración, anexión) (Figura 2A) y divisiones por procesos de fisión (sinónimos: división, fragmentación, esa cisión, desunión) (Figura 2B). Las mitocondrias derivan de eubacterias endosimbiontes capaces de realizar respiración aeróbica; este hallazgo fue propuesto por Merezhkovsky en 1905 y retomado por Margulis en 19675.
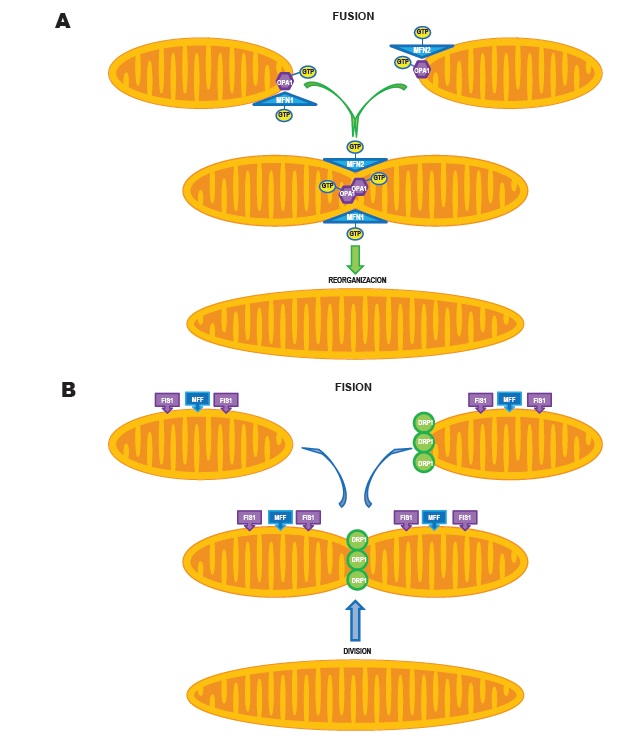
Figura 2. A. Fusión mitocondrial: se muestra esquemáticamente como las mitocondrias se unen a través del proceso de fusión, mediado por las actividades coordinadas de las mitofusinas 1 y 2 (MFN 1 y 2) en la membrana mitocondrial externa y de la proteína de la atrofi aóptica 1 (OPA1) en la membrana mitocondrial interna. Cuando las mitocondrias se fusionan, el contenido de sus matrices mitocondriales se entremezclan, creando organelas alargadas de color amarillo. Se muestra cómo las dos isoformas de mitofusina forman homodímeros o heterodímeros que conectan a las mitocondrias adyacentes, promoviendo la fusión. B. Fisión mitocondrial: se muestra esquemáticamente como se produce la fi sión, cuando la proteína relacionada con la dinamina-1 (DRP1) se activa y se mueve desde el citosol a la membrana mitocondrial externa. El montaje del aparato de fi sión es guiado por moléculas específi cas en la membrana mitocondrial externa (factor de fi sión mitocondrial [MFF] y la proteína de fi sión mitocondrial 1 [FIS1]), agrupándose en un microambiente por el contacto con el retículo endoplasmático. GTP: guanosín trifosfato.
El proceso de endosimbiosis seriada o teoría endosimbiótica describe la aparición de las células eucariotas o eucariogénesis como consecuencia de la sucesiva incorporación simbiogenética de diferentes bacterias de vida libre (procariotas). Este proceso fue propuesto por Lynn Margulis en diferentes artículos y libros: On the origin of mitosing cells (1967)6, Origins of eukaryotic cells (1975)7 y Symbiosis in cell evolution (1981)8, llegándose a conocer por el acrónimo inglés: SET (serial endosymbiosis theory). En la actualidad, se acepta que las eucariotas surgieron como consecuencia de los procesos simbiogenéticos descritos por Margulis, y así ha quedado demostrado el origen simbiogenético de las mitocondrias y los cloroplastos de los eucariontes6-9.
Historia
El descubrimiento de las mitocondrias fue un hecho colectivo. El gran número de términos que se refieren a esta organela es prueba de ello: blefaroplasto, condrioconto, condriómitos, condrioplastos, condriosomas, condriosferas, fila, gránulos fucsinofílicos, korner, fadenkörper, mitogel, cuerpos parabasales, vermículas, sarcosomas, cuerpos intersticiales, plasmosomas, plastocondrios, bioblastos. En 1918, en un trabajo luego citado por Lehninger, Cowdry intentó, sistematizar y unificar todos los términos4.
Probablemente, las primeras observaciones se deben al botánico suizo Kolliker, quien en 1880-1888 anotó la presencia de unos gránulos en células musculares de insectos a los que denominó sarcosomas, llegando a la conclusión de que presentaban membrana10. En 1882, el alemán Walther Flemming descubrió una serie de inclusiones a las que denominó fila11. En 1884 también fueron observados por Richard Altmann, quien más tarde en su obra publicada en Leipzig: "Die Elementar organismen",
describe una serie de corpúsculos que observa mediante una tinción especial que incluye fucsina. Especula que se trata de una suerte de parásitos independientes, con su propio metabolismo y los denomina bioblastos. El hallazgo fue rechazado como un artefacto de la preparación, y sólo más tarde fue reconocido como mitocondrias por N.H. Cowdry (1916)12.
Sin embargo, el nombre de mitocondria, que es el que alcanzó mayor fortuna, se debe a Carl Benda, quien en 189813 denominó así a unos gránulos que aparecían con gran brillo en tinciones de cristal violeta y alizarina, y que anteriormente habían sido denominados citomicrosomas por Velette St. George12. Estos gránulos observados por Benda con características morfológicas heterogéneas, a veces en forma de esfera y otras veces alargada, inspiraron el nombre de mitocondria (del griego mythos: relato, cuento, y chondrion: gránulo)14. En 1913, Otto Heinrich Warburg descubre la asociación de mitocondrias con enzimas de la cadena respiratoria, aunque ya Kingsbury (en 1912) había relacionado estos orgánulos con la respiración celular. En 1934, fueron aisladas por primera vez a partir de homogeneizados de hígado y, en 1948, Hogeboon, Schneider y Palade establecen definitivamente a la mitocondria como el lugar donde se produce la respiración celular15.
La presencia del ADN mitocondrial fue descubierta por Margit M. K. Nass y Sylvan Nass en 196310,16.
Dinámica mitocondrial
En 1914, las observaciones de Lewis establecieron el campo de la dinámica mitocondrial. Señalaron que "cualquier tipo de mitocondria tales como un gránulo, varilla o hilo puede a veces cambiar en cualquier otro tipo o puede fusionarse con otra mitocondria, o puede dividirse en otra o varias mitocondrias"17. El secreto de la vida dinámica de las mitocondrias fue revelado por el microscopio confocal: microscopio que emplea una técnica óptica de imagen para incrementar el contraste y/o reconstruir imágenes tridimensionales, utilizando un "pinhole" (agujero de alfiler) espacial (colimador de orificio delimitante) para eliminar la luz desenfocada o destellos de la lente en especímenes que son más gruesos que el plano focal18 (Figura 3).
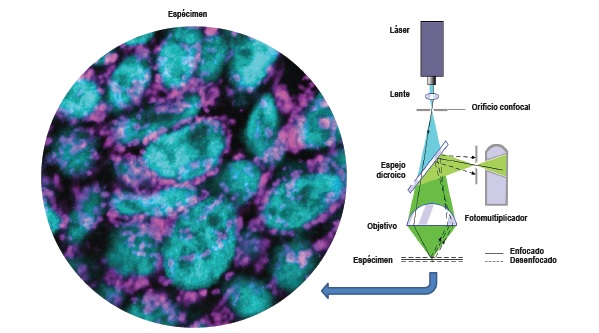
Figura 3. Esquema simplificado del funcionamiento de un microscopio confocal láser: muestra como el láser (luz azul) es filtrado por un orificio y un espejo dicroico; luego es enfocado mediante un lente objetivo sobre el espécimen y estimula la fluorescencia presente en el mismo (luz verde). La fluorescencia es recolectada por el objetivo y dirigida al espejo dicroico que la refleja y dirige hacia un multiplicador. Un segundo filtro con orificio se coloca frente al multiplicador y sólo deja pasar la luz proveniente del plano de enfoque (línea continua). La fluorescencia fuera de foco de las zonas que están por encima y por debajo del plano de enfoque (líneas discontinuas) no pasa por el orificio, no formando parte de la imagen.
La mayoría de las proteínas mitocondriales, incluyendo todas las involucradas en la fisión y la fusión, están codificadas por genes nucleares19.
El genoma mitocondrial (ADN mitocondrial, ADNmt/ ADNm o mtDNA/mDNA) es el material genético de las mitocondrias4. El ADN mitocondrial se reproduce por sí mismo semi-autónomamente cuando la célula eucariota se divide5.
Se ha demostrado que mutaciones en 228 genes nucleares y en 13 genes mitocondriales causan raros síndromes monogénicos en los que la disfunción mitocondrial juega un papel central en su patogénesis. Ejemplos de estos síndromes incluyen el síndrome MELAS (encefalomiopatía mitocondrial, acidosis láctica y episodios similares al accidente cerebrovascular isquémico causados por la mutación de la transferencia mitocondrial de los ARN), y la enfermedad de Leigh (causada por mutaciones en genes relacionados con la fosforilación oxidativa)19 (Figura 4). Y así aparecen los desordenes de la estructura mitocondrial como mecanismos de la enfermedad. Aunque algunos desórdenes de la dinámica mitocondrial son el resultado de la mutación monogénica, la mayoría de los cambios reflejados en la función o actividad de los mediadores de la fisión y la fusión son provocados por cambios en el ambiente celular. Hay un creciente reconocimiento respecto de que los desordenes dinámicos mitocondriales contribuyen a la patogénesis de enfermedades complejas, no consideradas clásicamente como de implicancia mitocondrial. Estas enfermedades incluyen: cánceres, enfermedades cardiovasculares y enfermedades neurodegenerativas. Recientemente se han identificado mediadores moleculares de la dinámica mitocondrial y se ha identificado su regulación postranslacional por una extensa red de quinasas, fosfatasas y mediadores de ubiquitinación que ofrecen una nueva comprensión de la biología celular y de nuevos objetivos terapéuticos. La fina sintonización de la fisión y fusión celular es fundamental para los procesos celulares tales como la homeostasis del calcio, la generación de ATP y las moléculas denominadas especies reactivas de oxígeno (ERO o ROS: reactive oxygen species, incluyen iones de oxígeno, radicales libres y peróxidos), que se encuentran reguladas por proteínas de la mitocondria que participan principalmente en la fusión mitocondrial. Teniendo, en consecuencia, un importante papel en la progresión del ciclo celular, la apoptosis, la mitofagia (degradación de unidades mitocondriales; forma especializada de autofagia por la cual se degradan y reciclan selectivamente las mitocondrias transportándolas al compartimento hidrolítico de la célula), y proteínas sensores del nivel de estrés celular y sensores de oxígeno (regulan el estado redox y controlan actividades enzimáticas, de canales iónicos y factores de transcripción).
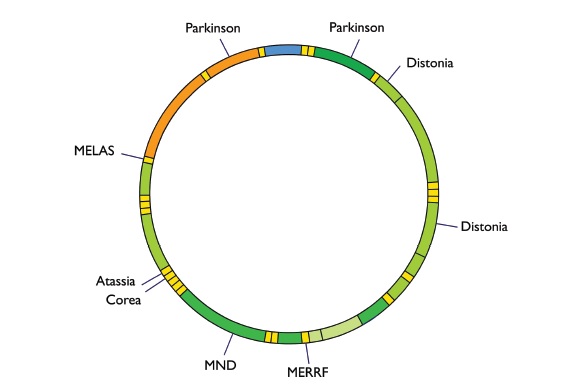
Figura 4. Representación del ADN mitocondrial mostrando los loci afectados en algunas enfermedades. MND: enfermedad de la motoneurona o esclerosis lateral amiotrófica. MELAS: encefalomiopatía mitocondrial, acidosis láctica y episodios parecidos a un accidente cerebrovascular. MERRF (myoclonic epilepsy with ragged red fibers): rara enfermedad mitocondrial que cursa principalmente con mioclonías y epilepsia.
El estado o reacción redox (reducción-oxidación) es toda reacción química en la que uno o más electrones se transfieren entre los reactivos, provocando un cambio en sus estados de oxidación. Para que exista una reacción de reducción-oxidación, en el sistema debe haber un elemento que ceda electrones, y otro que los acepte20.
Aunque la generación de ATP es la función principal de las mitocondrias (Figura 5 y Tabla 1), gran parte del impacto de la dinámica mitocondrial se refiere a efectos de la estructura sobre las capacidades no bioenergéticas de las organelas. Cinco funciones mitocondriales no canónicas (secundarias) están involucradas en el centro de la dinámica (forma y función) de las mitocondrias (Tabla 2). Estas relaciones son bidireccionales, lo que significa que tanto alteran como son alteradas por la dinámica mitocondrial: En primer lugar, las mitocondrias están relacionadas con el retículo endoplasmático en regiones especializadas de la adherencia de la membrana llamadas: membranas asociadas a las mitocondrias, que facilitar el flujo de calcio dentro de las mitocondrias. Las membranas del retículo endoplasmático asociadas a las mitocondrias permiten el aumento localizado en los niveles de calcio, propagándose a través de la célula en forma de onda21. La conexión retículo endoplasmático-mitocondrias tiene implicancias en el metabolismo y la homeostasis del calcio. El acoplamiento de estas organelas aumenta los niveles de calcio mitocondrial, pudiendo iniciar la apoptosis22 o, a niveles fisiológicos, mejorar el metabolismo oxidativo mediante la activación de la piruvato dehidrogenasa (PDH)23. Esta conectividad retículo endoplasmático-mitocondrias está regulada por la mitofusina-2, pudiendo crear microdominios (plataformas estructurales de lípidos y proteínas que propician la eficiente modulación de los procesos fisiológicos asociados a la membrana) que facilitan la fisión24,25 (Figura 2B y Figura 5).
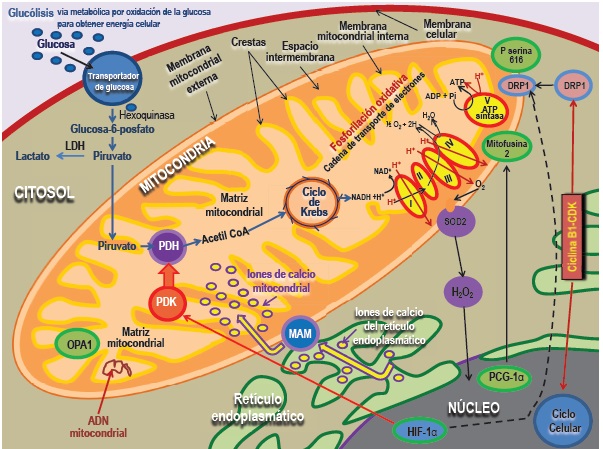
Figura 5. Dinámica y metabolismo mitocondrial. La membrana mitocondrial externa es permeable y la membrana mitocondrial interna es impermeable. Ambas están separadas por un espacio intermembrana dentro del cual el ión hidrógeno (H+) es bombeado durante el transporte de electrones. Dentro de la membrana mitocondrial interna existe una matriz mitocondrial que contiene el ADN mitocondrial, proteínas y ribosomas. Dentro de las crestas residen los cuatro megacomplejos de la cadena de transporte de electrones y la ATP-sintasa, donde el adenosín difosfato (ADP) y el fosfato inorgánico (Pi) se unen para producir adenosín trifosfato (ATP). El piruvato generado en el citosol por la glucólisis (vía metabólica por oxidación de la glucosa para obtener energía celular) entra en la mitocondria y genera la energía celular o permanece en el citosol, convirtiéndose en lactato por la lactato deshidrogenasa (LDH) si el metabolismo mitocondrial es inhibido. Si el piruvato sigue la primera vía, una vez dentro de la mitocondria, se convierte en acetil coenzima A (CoA) por la piruvato deshidrogenasa (PDH), principal regulador del metabolismo oxidativo. La PDH puede ser inhibida por la enzima piruvato deshidrogenasa quinasa (PDK) o activarse por aumento del calcio mitocondrial. El acetil Co A genera combustible en el ciclo de Krebs y dadores de electrones (NADH: nicotín adenin dinucleótido reducido y FAD: dinucleótido de fl avina y adenina), pasando a la cadena de transporte de electrones para reducir oxígeno molecular. Este fl ujo de electrones genera el gradiente de iones de hidrógeno quimiosmótico que alimenta la síntesis de ATP. Además, el fl ujo de electrones genera especies reactivas de oxígeno (ERO o ROS), tales como el anión superóxido (O2-), que son convertidos en moléculas de peróxido de hidrógeno (H2O2) por la superóxido dismutasa 2 (SOD2). Los mediadores de la fusión (mitofusina-1 y mitofusina-2) y la enzima que genera lípidos fusogénicos, la fosfolipasa D, residen en la membrana mitocondrial externa, mientras que la proteína de la atrofi a óptica 1 (OPA1) se encuentra en la membrana mitocondrial interna. La proteína relacionada con la dinamina-1 (DRP1) mediadora de la fi sión se encuentra en el citosol, pero cuando es activada, como por ejemplo después de la fosforilación (P) de la serina 616, se ubica en la membrana mitocondrial externa. El ciclo celular y la fi sión mitocondrial están vinculados por la quinasa ciclina B1-dependiente de ciclina (CDK), asegurando la coordinación de la división nuclear y mitocondrial. El receptor activado por proliferadores de peroxisoma y coactivador 1α (PGC-1α) y el factor inducible por hipoxia 1 α (HIF-1 α) proporcionan el control transcripcional de la fi sión y de la fusión. El calcio se mueve desde el retículo endoplasmático a las mitocondrias a través de zonas de adherencia a la membrana llamadas membranas del retículo endoplasmático asociadas a mitocondrias (MAM). Los niveles de calcio mitocondrial regulan la actividad de la PDH y la apoptosis. La mitocondria y el retículo endoplasmático también interactúan para crear microambientes que dirigen la fi sión. Modifi cado de Archer SL. N Engl J Med 2013;369:2236-2251.
Tabla 1. Funciones de la mitocondria

Tabla 2. Funciones secundarias (no canónicas) de las mitocondrias

En segundo lugar, la biogénesis mitocondrial regulan la cantidad de mitocondrias para satisfacer la demanda de energía de la célula y compensar el daño celular. Este proceso está mediado por el gen coactivador 1α del receptor que activa la proliferación de peroxisomas γ (PGC-1α), el cual es importante para la dinámica mitocondrial, ya que es un coactivador transcripcional de la fusión del mediador mitofusina-226,27.
En tercer lugar, las mitocondrias tienen un programa de control de calidad llamado: mitofagia. La mitofagia mantiene la salud celular a través de un proceso de degradación selectiva de la mitocondria ante un daño en esta organela, despolarizando a las mitocondria en vacuolas autofágicas para su eliminación a través de los lisosomas28,29. La fisión separa a mitocondrias despolarizadas, mientras coordina la disminución de la regulación de los mediadores de fusión, impidiendo el trabajo de reintegración y facilitando de este modo la mitofagia. Fallos en este proceso están relacionados con enfermedades como el síndrome de Parkinson30,31.
En cuarto lugar, la actividad mitocondrial atraviesan activamente el citosol mediante la acción de la dineína y la kinesina (importantes proteínas motoras asociadas a los microtúbulos para el transporte intracelular)32. Es incierto si la movilidad y dinámica mitocondrial tienen una relación obligatoria; sin embargo, la dineína (motor molecular para el transporte mitocondrial) regula, también, la fisión33. En quinto lugar, las mitocondrias son los sensores de oxígeno en las células dentro del sistema de detección de oxígeno homeostático, como las células musculares lisas de la arteria pulmonar y del conducto arterioso persistente34. Estas mitocondrias especializadas varían la producción de la difusión de oxígeno reactivo por parte de la cadena de transporte del electrón en proporción a los niveles de oxígeno celular, permitiendo la regulación redox de los canales iónicos, enzimas y factores de transcripción34. La dinámica mitocondrial es el primer paso en los mecanismos de señalización redox35,36.
La fisión crea mitocondrias más pequeñas, diminutas, que, dependiendo del contexto, son más capaces de generar especies de oxígeno reactivas, facilitando la mitofagia y acelerando la proliferación celular. La fusión produce una red mitocondrial más interconectada, que mejora la comunicación con el retículo endoplasmático. La fusión también permite la difusión de contenido de la matriz entre las mitocondrias (Figura 2A), diluyendo la mutación acumulada del ADN mitocondrial37 y proteínas oxidadas.
Tanto la fi sión como la fusión son mediadas por un pequeño número de enzimas guanosín-trifosfatasas (GTPasas) altamente conservadas38,39 (Figura 2A y 2B). La fisión es mediada por la proteína relacionada con la dinamina-1 (DRP1: dynamin-related protein 1)40,41, una proteína citosólica que al activarse se transloca a la membrana mitocondrial externa (Figura 5). Así, la multimerización de la DRP1 crea una estructura en forma de anillo que se contrae y divide a la organela42,43 (Figura 2B). La DRP1 se dirige activamente a la membrana mitocondrial externa a través de proteínas receptoras no GTPasas, tales como la proteína de fisión mitocondrial 1 (FIS1)43, el factor de fisión mitocondrial (MFF)44 y el factor de elongación mitocondrial 145 (Figura 2B). El ensamble de los aparatos de fisión es asistido por el retículo endoplasmático, que hace contacto con la mitocondria, creando un microdominio para el ensamble de DRP1, MFF y proteínas proapoptóticas46.
La actividad de la DRP1 se regula rápidamente por efectos opuestos de la fosforilación en dos llaves serinas. La fosforilación de la serina 616 aumenta la actividad del DRP1, mientras que la fosforilación de la serina 637 la reduce (Figura 5). Cada serina es el blanco de diferente quinasas y fosfatasas, vinculando de esta manera la fisión mitocondrial a procesos celulares cruciales. La relación de la fosforilación de la serina 616 a serina 637 determina la actividad DRP1 y refleja los efectos agregados de muchas quinasas y fosfatasas.
La fusión está mediada por las isoformas mitofusina 1 y mitofusina 2 en la membrana mitocondrial externa y por la proteína de la atrofia óptica 1 (OPA1) en la membrana mitocondrial interna47 (Figura 2A y Figura 5). Las mitofusinas 1 y 2 están enfocadas a la mitocondria por secuencias en sus dominios transmembrana y C-terminal48. Las mitofusinas inician la fusión mediante la creación entre mitocondrias adyacentes de homodimérica o heterodimérica, antiparalela y vínculos de doble espiral49 (Figura 2A). La mitofusina-2 está también localizada en el retículo endoplasmático, donde altera las características morfológicas y promueve el anclaje retículo endoplasmático-mitocondrial48, y así mejorar la captación de calcio mitocondrial24. La OPA1 tiene ocho variantes de empalme, cada una con una diferente actividad de fusión y susceptibilidad de la proteasa mitocondrial50. Los mediadores de fusión son regulados por el metabolismo mitocondrial, y cuando están disminuyendo o disfuncionando por lo general hay una reducción de la capacidad oxidativa mitocondrial50. La fusión y la fisión están guiadas por lípidos generados por la fosfolipasa mitocondrial D, especialmente el acido fosfatídico25. El grupo de cabeza lipídico del ácido fosfatídico (negativamente cargado) provoca una curvatura negativa de bicapas de lípidos y recluta proteínas adaptadoras, promoviendo la fusión25. Sin embargo, el ácido fosfatídico puede ser hidrolizado por la lipina-1, creando diacilglicerol, que promueve la fisión51.
Alteraciones mitocondriales y su relevancia clínica
Podemos considerar tres mecanismos de alteración mitocondrial:
A. Un mecanismo preponderantemente estructural donde el inter-juego de fusión y fisión mitocondrial se ve alterado. Parecería cada vez más evidente la validez de la "teoría simbiótica de la mitocondria", que postula que esta organela fue en algún momento de la evolución una bacteria que en la dinámica de la filogenia pasó a formar parte de la célula eucariota. La mitocondria puede adquirir estructura granular o filamentosa, dividirse y fusionarse nuevamente con distintas partes, regulado por señales y moduladores proteicos de su membrana, o bien fusionarse con otra mitocondria presente en la misma célula. Este proceso de fusión y fisión alcanza su máxima complejidad en la célula humana. A los fines de la transferencia de conocimientos de la investigación del laboratorio a la clínica, resulta preponderante los disturbios en la dinámica mitocondrial que puede dar lugar a distintas enfermedades; particularmente: persistencia del ductus arterioso, hipertensión arterial pulmonar (HAP) y cáncer de pulmón52,53.
B. Alteraciones de la maquinaria bioenergética, como la disminución del proceso oxidativo a partir de la inhibición de la enzima piruvato deshidrogenasa (PDH) -enzima reguladora del paso de piruvato (metabolito final de la vía de degradación de la hexosa-monofosfato) a acetil coenzima A (acetil-coA) -que junto con el oxalacetato dan lugar al ciclo de Krebs (la acetil-coA es el combustible; el oxalacetato la chispa encendedora) resultando en la generación de ATP y oxidación de NADH con la consiguiente transferencia de H+ a la cadena respiratoria con el resultado final de producción de CO2, H2O y O2, que por la enzima super-óxido de dismutasa (SOD) localizada en las membrana mitocondrial se transforma en H2O2 (ROS), difundiendo hacia el núcleo (Figura 5). Alteraciones en estos pasos pueden generar HAP, ductus arterioso persistente y cáncer de pulmón.
C. Participación en la respuesta inflamatoria sistémica (RIS), el disparador ("trigger") es la unión de la organela inflamasoma RPL3 -la membrana más interna del retículo granular en reposo- que por un mecanismo no conocido adopta una conformación circular y se traslada a la membrana externa de la mitocondria, produciendo el estímulo para amplificar la formación de ROS y consiguiente estimulación del endotelio vecino a la célula "blanco" con la síntesis y liberación de citoquinas pro-inflamatorias y otros mediadores que perpetuarán la RIS presente entre otros procesos mórbidos en aterosclerosis, diabetes mellitus y cáncer, con las secuelas clínicas de fatiga, malnutrición, hipotensión ortostática y estado de hipercoagulabilidad; el marcador bioquímico de mayor sensibilidad a la fecha es la proteína C reactiva que se encuentra elevada54,55.
Enfermedades cardiovasculares que involucran a la dinámica mitocondrial
Los trastornos de la dinámica mitocondrial están implicados en enfermedades neurodegenerativas, neoplásicas, endocrinas (diabetes) y cardiovasculares (hipertensión pulmonar, ductus arterioso persistente, etc.) (Figura 4).
Este mismo patrón de desorden de la proliferación celular y resistencia a la apoptosis es vista en enfermedades como el cáncer y enfermedades cardiovasculares. La disfunción mitocondrial también es vista en enfermedades neurodegenerativas donde los niveles de fisión, se hayan incrementados y hay una alteración de la fusión resultando en fragmentación mitocondrial como la regla en las enfermedades neurodegenerativas de los adultos. Entre ellas encontramos al parkinsonismo familiar, la enfermedad de Alzheimer, la enfermedad de Huntington y la esclerosis lateral amiotrófica, como así también neuropatías como la enfermedad de Charcot-Marie-Tooth y la atrofia óptica30,31.
Hipertensión arterial pulmonar
La hipertensión arterial pulmonar (HAP) se define como un grupo de enfermedades caracterizadas por el aumento progresivo de la resistencia vascular pulmonar (RVP) que conduce al fallo del ventrículo derecho y a la muerte prematura56-58. Es una vasculopatía pulmonar obstructiva; aunque la vasoconstricción, la inflamación y la trombosis contribuyen a la patogénesis, una "visión oncológica" está emergiendo, la cual sostiene que una proliferación excesiva y una deficiente apoptosis participan en la progresión de la enfermedad59.
Muchos factores contribuyen al fenotipo neoplásico de la HAP60,61. Tanto el sensado de oxígeno como la disfunción mitocondrial son desórdenes observados en la célula muscular lisa de la arteria pulmonar, como se indica por la activación normal del oxígeno del factor 1α inducible por hipoxia (HIF-1α) y la fragmentación mitocondrial62.
La huella de la dinámica mitocondrial en la HAP es similar a la hallada en el cáncer, con disminución de la fusión26 mediada por la mitofusina-2 y aumento excesivo de la fisión mediada por la DRP136.
La HAP resulta y es, a su vez, un modelo clínico del desbalance entre los procesos de fisión y fusión mitocondrial con un predominio del primero sobre el segundo; la molécula clave que regula la fisión mitocondrial es la DRP1. Esta proteína se encuentra inactiva durante la fusión mitocondrial por prevalencia de la fosforilación del residuo 687 de la serina (ser687) respecto al residuo ser616, y una activación de las proteínas de membrana mitofusina 1 y 2, sobre todo este último subtipo. Al incrementarse la fisión mitocondrial se produce un desbalance de la fosforilación a favor de ser616 y disminución de la mitofusina 2; la preponderancia de la fisión mitocondrial trae aparejado alteraciones a nivel del circuito pulmonar: yendo esta vez desde la clínica a lo molecular se correlaciona la reducción en la longitud de los troncos de los vasos pulmonares en forma de corte abrupto de sus extremidades y mayor señal en sus ramas también amputadas en una resonancia magnética nuclear, donde también se evidencia agrandamiento y aumento de la señal del ventrículo derecho, la pérdida de vasos pulmonares se correlaciona a nivel histológico por obstrucción arterial de tipo plexiforme, con proliferación de la íntima, la patente molecular es la de una promoción de los mecanismos anti-apoptóticos en la capa muscular arterial; la principal molécula implicada es el factor inductor de hipoxia 1 alfa (HIF-1alfa: hypoxia-inducible factor 1α). El HIF-1alfa despliega múltiples acciones: se dirige desde el núcleo a la mitocondria, donde estimula a la enzima piruvato deshidrogenasa kinasa (PDK) que inhibe a la PDH; por lo tanto disminuye el mecanismo oxidativo de la mitocondria; perpetúa el desbalance de DRP1/mitofusina-2, con la consiguiente perpetuación del predominio del proceso de fisión sobre el de fusión mitocondrial (Figura 5); a su vez, estimula la expresión por el receptor del factor de crecimiento endotelial vascular 1 y 2 (VEGFR1-2: vascular endothelial growth factor receptor 1-2) por dos vías: una por estimulación directa, y otra indirecta a través de la inducción de la enzima ciclooxigenasa-2 (COX-2), que también estimula la expresión del VEGFR1-2, además de sintetizar prostaglandina E2 (PG-E2) a partir del ácido araquidónico de la membrana interna del endotelio, la PG-E2 es un mediador junto con las citoquinas en la perpetuación de la RIS, que en la HAP está presente de una manera baja y persistente63,64. El desencadenamiento del accionar HIF1- VEGF1-2, producen los vasos de la periferia de los troncos pulmonares con caracteres de sinuosidad, amputación y un patrón propio de respuesta a aminas vasoactivas. A nivel molecular se produce la activación del oncogen c-myc que perpetúa la proliferación de la íntima e hiperplasia de la capa muscular, la activación del oncogen c-myc induce triple respuesta: anti-apoptosis, crecimiento de las capas celulares y remodelación de los vasos36,65.
Ductus arterioso persistente
Minutos posteriores al nacimiento se produce una disminución marcada del flujo sanguíneo por el ductus arterioso persistente (DAP) como consecuencia tanto de la caída de la secreción por la placenta de PGI2, como por la vasoconstricción del DAP debido al incremento de la PO2 por la respiración pulmonar; en el 90% de los neonatos el DAP se cierra a las 72 horas por estos mecanismos, produciéndose una reacción fibrosa que terminará por ocluirlo totalmente en promedio a la semana de vida extra-uterina66,67. A nivel molecular la explicación del cierre del DAP se explica porque el mecanismo de la permeabilidad del conducto es subyacente al mecanismo sensor de oxígeno por el potencial redox de la mitocondria, las ROS de la mitocondria son convertidas en la membrana en H2O2 por la SOD, el ión OH- regula los canales iónicos de la célula, que a su vez está en equilibrio con la formación de la DRP1; tanto la regulación de los canales iónicos, como la cantidad de DRP1 están en conexión con la fisión mitocondrial, si se inhibe la misma el DAP permanecerá permeable, ya que habrá menor potencial de óxido-reducción y DRP1, y por ende menor formación de OH-. Vale decir que la estabilidad mitocondrial es determinante para el estado tanto de los vasos arteriales pulmonares como del conducto arterioso, y su afectación, ya sea en más o en menos, tendrá consecuencias nocivas para el organismo. Por otro lado el oxígeno induce el cierre del DAP por estimulación de la ciclina B1-CDK1, proteína reguladora del ciclo celular modulada por la fisión mitocondrial que promueve la transición de las células musculares del ductos de la fase G2 a M, con hiperplasia y posterior aposición de material conectivo con el cierre del mismo38,52.
Diabetes mellitus
Los defectos de fusión mitocondrial promueven el desarrollo de diabetes en modelos murinos de diabetes mellitus tipo 1. En pacientes con obesidad o diabetes mellitus tipo 2, la fusión se ve afectada también, como evidencia de la disminución de la expresión de mitofusina 2 y del tamaño mitocondrial68. La supresión hepática de mitofusina 2 incrementa la generación de especies reactivas de oxígeno y provoca intolerancia a la glucosa por disminución de la secreción de insulina69. La disminución de mitofusina 2 impide el consumo de oxígeno y promueve la diabetes. Ratones con menores porcentajes de células pancreáticas β presentan una alteración de la secreción de insulina estimulada por la glucosa, desarrollando hiperglucemia70. Aunque el grave deterioro de la fusión promueve diabetes, es poco probable que la restauración de la fusión mitocondrial sea sencilla, ya que la fragmentación mitocondrial es requerida para la mitofagia. Las mitocondrias, bajo fisión asimétrica, resultan tanto en mitocondrias normales como despolarizadas, mitocondrias disfuncionales deficientes en OPA171. La deficiencia aislada de OPA1 de mitocondrias anormales evita su reintegración en cadena. Este deterioro de la fusión localizada y mitofagia es beneficioso. De hecho, la inhibición de la mitofagia por una mejora de la fusión (como sobreexpresión de la OPA1) conduce a la acumulación de mitocondrias dañadas con alteración de la respiración y reducción de la secreción de insulina71.
Isquemia e injuria de reperfusión
A pesar de las eficaces técnicas de reanimación, la mortalidad de un paro cardíaco sigue siendo alta, en parte a causa de la isquemia vascular e injuria de reperfusión.
En el riñón, el mdivi-1 (mitochondrial fission inhibitor: inhibidor selectivo de la fisión mitocondrial) protege a las células tubulares renales de la fisión inducida por la lesión por isquemia-reperfusión72; también fue vista su acción protectora en un modelo de lesión de isquemiareperfusión cardiaca73.
Durante el paro cardíaco, la DRP1 se activa mediante la desfosforilación de DRP1 mediada por la calcineurina en serina 63774. La resultante fisión aumenta la producción de especies reactivas del oxígeno, aumenta los niveles de calcio y limita la relajación diastólica. La inhibición de la DRP1, ya sea directamente o mediada por la calcineurina, puede tener un efecto terapéutico.
Miocardiopatía
La ablación cardíaca condicional de isoformas de mitofusina simultáneamente tiene efectos perjudiciales sobre las características morfológicas mitocondriales, la respiración y la función contráctil, lo que resulta en la muerte por falla cardíaca75,76. Por el contrario, la deleción (anomalía estructural) de una sola isoforma de mitofusina es cardioprotectora contra la isquemia-injuria de reperfusión y de especies reactivas de oxígeno. La supresión cardíaca de mitofusina-2 da como resultado hipertrofia y disfunción mitocondrial en forma leve, con una ligera reducción en la función de ventrículo izquierdo. Sin embargo, en estos ratones experimentales se ha reducido la susceptibilidad a la activación del poro de transición de permeabilidad mitocondrial77. Posteriormente, los mismos investigadores mostraron que la supresión condicional de mitofusina-1 también produce una función cardíaca basal normal y se encuentran protegidos de la cardiotoxicidad inducida por especies de oxígeno reactivo.
Similitudes con el cáncer
La disfunción mitocondrial descripta en enfermedades vasculares como la HAP y el DAP comparte mecanismos íntimos comunes con el cáncer de pulmón. Se conoce que las características de las células tumorales son un estado anti-apoptótico, con remodelación de los vasos del microambiente (angiogénesis) y crecimiento del tejido patológico. Hacia 1930, el bioquímico alemán Otto Heinrich Warburg describió el fenómeno que lleva su nombre, por el cual la célula tumoral, independientemente de la concentración de oxígeno del medio, utiliza siempre el mismo porcentaje, con menor actividad de la PDH en la vía oxidativa de la glucosa, y una mayor actividad a nivel citoplasmático de la láctico deshidrogenasa (LDH), lo cual genera ácido láctico a partir del piruvato, con generación de 2 ATP en lugar de 36 ATP, la provisión de H+ a la cadena respiratoria mitocondrial se hacía vía el ciclo de las pentosas, o sea que vías triviales para una célula normal, no lo son para la célula neoplásica. Warburg consideró que la mitocondria de la célula tumoral era impermeable y por ello no se podía continuar la oxidación aeróbica de la glucosa en la misma. El mecanismo se comprobó por otros investigadores.
En 1931 fue galardonado con el Premio Nobel de Medicina, por su descubrimiento de la naturaleza y el modo de acción de la vía oxidativa aeróbica de la glucosa, junto a Embden y Meyerhof. En años recientes se comprobó que la membrana externa de la mitocondria, no es impermeable como pensaba Warburg, y el fenómeno que el describió constituye un fenotipo bioquímico de la célula tumoral78,79. El advenimiento del conocimiento acerca de la disfunción mitocondrial posibilitó comprender que ciertos fenómenos que acontecen en la célula tumoral son comunes a alteraciones cardiovasculares y que no sólo el efecto Warburg estaba implicado en el metabolismo tumoral. Así, tanto la HAP como el DAP comparten con el cáncer de pulmón alteraciones en la DRP1 que posibilitan un desbalance a favor de la fisión mitocondrial con menor fusión, activación de la ciclina B1-CDK1 con progresión en el ciclo celular y división de la células tumorales, más estimulación del HIF-1alfa con su acción sobre COX-2 y VEGFR1-2 en el remodelado de los vasos del microambiente tumoral. A su vez, se ha comprobado que la disminución de la mitofusina 2, que estimula la fusión mitocondrial, activa mecanismos de reproducción tumoral e hipertrofia del miocardio con disfunción ventricular izquierda52,76.
De este modo procesos patológicos que hasta hace unos años se consideraban antagónicos como los cardiovasculares y los neoplásicos, comparten mecanismos en común, determinando que algunos padezcan una u otra enfermedad (sin ser excluyentes) es un desafío abierto.
Conclusiones
La disfunción mitocondrial está involucrada en los mecanismos de una gran variedad de enfermedades, pudiendo ofrecer objetivos terapéuticos comunes. Antes de intentar utilizar la dinámica mitocondrial terapéuticamente, se requiere una evaluación adicional para identificar los objetivos moleculares óptimos y definir las dosis seguras y eficaces de moduladores de fisión y fusión, eligiendo selectivamente las poblaciones celulares involucradas y requiriéndose moduladores moleculares o farmacológicos adicionales de fisión y fusión.
El desarrollo de tratamientos metabólicos y farmacológicos eficaces para enfermedades mitocondriales definidas es una promesa para futuras terapias que podrían mejorar los síntomas de una amplia gama de alteraciones metabólicas y trastornos degenerativos e incluso el envejecimiento. Siendo necesarias futuras líneas de investigación para definir las conductas a seguir.
Recursos financieros
Los autores no recibieron ningún apoyo económico para la investigación.
Conflicto de intereses
Los autores declararon no tener conflicto de intereses.
1. Cerchietti L, Navigante A, Castro M. Effects of eicosapentaenoic and docosahexaenoic n-3 fatty acids from fish oil and preferential Cox-2 inhibition on systemic syndromes in patients with advanced lung cancer. Nutr Cancer 2007;59(1):14-20. [ Links ]
2. Navigante A, D'ortencio A. Insuficiencia cardíaca y respuesta inflamatoria sistémica. Insuf Card 2009;4(4):186-189. [ Links ]
3. Marín-García J, Goldenthal MJ. La mitocondria y el corazón. Rev Esp Cardiol 2002; 55(12):1293-1310. [ Links ]
4. Scheffler IE. Mitochondria. 2nd edition. Ed. John Wiley and Sons, Inc. Hoboken, New Jersey, 2008: 10-25. [ Links ]
5. Knoll AH, Lynn Margulis. 1938-2011. Proc Natl Acad Sci U S A 2012;109:1022. [ Links ]
6. Margulis L. On origen of mitosing cells. J Theoret Biol 1967;14 (3): 225. [ Links ]
7. Margulis L. Origins of eukaryotic cells. 1st edition. Yale University Press, New Haven, 1975: 26-48. [ Links ]
8. Margulis L. Symbiosis in cell evolution. Ed. Freeman, New York, 1981. [ Links ]
9. Margulis L, Sagan D. Marvellous microbes. Resurgence 2001; 206: 10-12. [ Links ]
10. Sastry KV, Singh SP, Tomar BS. Cell & developmental biology. Ed. Rastogi publications. New Delhi. India. 2006. ISBN 9788171336784. [ Links ]
11. Flemming W. Zellsubstanz, kern und zelltheilung. Ed. Vogel-Verlag KG. Leipzig. 1882. https://archive.org/details/ zellsubstanzker02flemgoog [ Links ]
12. Martin WF, Müller M. Origin of mitochondria and hydrogenosomes. Ed. Springer. Berlín. 2007. ISBN 9783540385011. [ Links ]
13. Benda, C. Ueber die Spermatogenese der Vertebraten und höherer Evertebraten, II. Theil: Die Histiogenese der Spermien (On spermatogenesis of vertebrates and higher invertebrates, Part II: The histogenesis of sperm). Archiv für Anatomie und Physiologie 1898;73:393-398. [ Links ]
14. O'Rourke B. From bioblasts to mitochondria: ever expanding roles of mitochondria in cell physiology. Front Physiol 2010;1:7. [ Links ]
15. Gupta PK. Genetics: classical to modern. Ed. Rastogi Publications, New Delhi, India. 2007. ISBN 9788171338962. [ Links ]
16. Nass MMK, Nass S. Intramitochondrial fibers with DNA characteristics. Fixation and electron staining reactions. J Cell Biol 1963; 19:593-611. [ Links ]
17. Lewis MR, Lewis WH. Mitochondria in tissue culture. Science 1914;39:330-3. [ Links ]
18. Pawley JB. Handbook of Biological Confocal Microscopy. 3rd edition. Ed. Springer. Berlin. 2006. ISBN 038725921X. [ Links ]
19. Koopman WJ, Willems PH, Smeitink JA. Monogenic mitochondrial disorders. N Engl J Med 2012;366(12):1132-41. [ Links ]
20. Mailloux RJ, Jin X, Willmore WG. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol 2014; 2: 123-139. [ Links ]
21. Patergnani S, Suski JM, Agnoletto C, et al. Calcium signaling around Mitochondria Associated Membranes (MAMs). Cell Commun Signal 2011;9:19. [ Links ]
22. Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ, Rizzuto R. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell 2004;16:59-68. [ Links ]
23. Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta 2009;1787:1309-16. [ Links ]
24. de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008;456(7222):605-10. [ Links ]
25. Yang CY, Frohman MA. Mitochondria: signaling with phosphatidic acid. Int J Biochem Cell Biol 2012;44:1346-50. [ Links ]
26. Ryan JJ, Marsboom G, Fang YH, et al. PGC1α-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am J Respir Crit Care Med 2013;187(8):865-78.
27. Zorzano A. Regulation of mitofusin-2 expression in skeletal muscle. Appl Physiol Nutr Metab 2009;34(3):433-9. [ Links ]
28. Tolkovsky AM. Mitophagy. Biochim Biophys Acta 2009;1793(9):1508-15. [ Links ]
29. Zhang J, Ney PA. Reticulocyte mitophagy: monitoring mitochondrial clearance in a mammalian model. Autophagy 2010;6(3):405-8. [ Links ]
30. Ryan BJ, Hoek S, Fon EA. Wade-Martins R. Mitochondrial dysfunction and mitophagy in Parkinson's: from familial to sporadic disease. Trends Biochem Sci 2015;40(4):200-210. [ Links ]
31. Deas E, Wood NW, Plun-Favreau H. Mitophagy and Parkinson's disease: The PINK1–parkin link. Biochim Biophys Acta 2011;1813(4): 623-633.
32. Wang X, Winter D, Ashrafi G, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011;147(4):8 93-906. [ Links ]
33. Varadi A, Johnson-Cadwell LI, Cirulli V, Yoon Y, Allan VJ, Rutter GA. Cytoplasmic dynein regulates the subcellular distribution of mitochondria by controlling the recruitment of the fission factor dynaminrelated protein-1. J Cell Sci 2004;117: 4389-400. [ Links ]
34. Weir EK, López-Barneo J, Buckler KJ, Archer SL. Acute oxygen-sensing mechanisms. N Engl J Med 2005;353(19):2042- 55. [ Links ]
35. Hong Z, Kutty S, Toth PT, et al. Role of dynamin-related protein 1 (Drp1)-mediated mitochondrial fission in oxygen sensing and constriction of the ductus arteriosus. Circ Res 2013;112:802-15. [ Links ]
36. Marsboom G, Toth PT, Ryan JJ, et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res 2012; 110(11):1484-97. [ Links ]
37. Santel A, Frank S, Gaume B, Herrler M, Youle RJ, Fuller MT. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J Cell Sci 2003;116:2763-74. [ Links ]
38. Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science 2012;337:1062-5. [ Links ]
39. Ishihara N, Otera H, Oka T, Mihara K. Regulation and physiologic functions of GTPases in mitochondrial fusion and fission in mammals. Antioxid Redox Signal 2013;19(4):389-99. [ Links ]
40. Cribbs JT, Strack S. Functional characterization of phosphorylation sites in dynamin-related protein 1. Methods Enzymol 2009;457:231-53. [ Links ]
41. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 2003;160(2):189-200. [ Links ]
42. Zhu PP, Patterson A, Stadler J, Seeburg DP, Sheng M, Blackstone C. Intra- and intermolecular domain interactions of the C-terminal GTPase effector domain of the multimeric dynamin-like GTPase Drp1. J Biol Chem 2004;279(34):35967- 74. [ Links ]
43. Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell 2004;15(11):5001- 11. [ Links ]
44. Otera H, Wang C, Cleland MM, et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 2010;191(6):1141-58. [ Links ]
45. Zhao J, Liu T, Jin S, et al. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J 2011;30(14):2762-78. [ Links ]
46. Hoppins S, Nunnari J. Cell biology: mitochondrial dynamics and apoptosis - the ER connection. Science 2012;337(6098): 1052-4. [ Links ]
47. Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A 2004;101(45):15927-32. [ Links ]
48. Rojo M, Legros F, Chateau D, Lombès A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrana GTPase Fzo. J Cell Sci 2002;115:1663-74. [ Links ]
49. Liesa M, Palacín M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev 2009;89(3):799- 845. [ Links ]
50. Zorzano A, Liesa M, Sebastián D, Segalés J, Palacín M. Mitochondrial fusión proteins: dual regulators of morphology and metabolism. Semin Cell Dev Biol 2010;21(6):566-74. [ Links ]
51. Huang H, Gao Q, Peng X, et al. piRNA-associated germline nuage formation and spermatogenesis require MitoPLD profusogenic mitochondrial-surface lipid signaling. Dev Cell 2011;20:376-87. [ Links ]
52. Archer SL. Mechanisms of disease. Mitocondrial dynamics. Mitochondrial fission and fusion in human diseases. N Engl J Med 2013; 369(11):2236-2251. [ Links ]
53. Kashatus D, Lim K, Brady D, et al. RALA and RALB1 regulate mitochondrial fission at mitosis. Moll Cell Biol 2011; 13(9):1108-15. [ Links ]
54. Navigante A, Morgado PC, Casbarien O, Delgado NL, Giglio R, Perman M. Relationship between weakness and phase angle in advanced cancer patients with fatigue. Support Care Cancer 2013;21(6):1685-1690. [ Links ]
55. Pepys M, Hirschfield G. C-reactive protein: a critical update. J Clin Invest 2003; 111(12):1805-12. [ Links ]
56. Simonneau G, Galie N, Rubin L, et al. Clinical classification of pulmonary arterial hypertension. J Am Coll Cardiol 2004;43(12): S5-12. [ Links ]
57. Galiè N, Torbicki A, Barst R, Dartevelle P, Haworth S, Higenbottam T, et al. Task Force. Guidelines on diagnosis and treatment of pulmonary arterial hypertension. The Task Force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of Cardiology. Eur Heart J 2004;25(24):2243-78. [ Links ]
58. Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension. Rev Esp Cardiol 2016;69(2):177.e1-e62. [ Links ]
59. Archer SL, Weir EK, Wilkins MR. Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation 2010; 121(18):2045-66. [ Links ]
60. Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol 2011;8:443-55. [ Links ]
61. Voelkel NF, Gomez-Arroyo J, Abbate A, Bogaard HJ, Nicolls MR. Pathobiology of pulmonary arterial hypertension and right ventricular failure. Eur Respir J 2012;40(6):1555-65. [ Links ]
62. Bonnet S, Michelakis ED, Porter CJ, et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 2006;113(22):2630-41. [ Links ]
63. Cerchietti L, Navigante A. Síndrome de disfunción inmunometabólico en cáncer. Actualización en Nutrición 2004;5(3):11-16. [ Links ]
64. González C. Caquexia, inflamación y cáncer: las piezas comienzan a encajar. Actualización en Nutrición 2004;5(3):8-10. [ Links ]
65. Berdasco M, Esteller M. Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev Cell 2010; 19(5): 698-711. [ Links ]
66. Grassi de Gende A. Circulación en lechos especiales. En: Cingolani H, Houssay A y col. Fisiología humana de Houssay. 7ma edición. Ed.: El Ateneo. Buenos Aires. 2005. Cap.29: 354-7. [ Links ]
67. Heng Z, Kutty S, Toth P, et al. Role of dynamin- related protein 1 (Drp1)- mediated mitochondrial fission in oxygen sensing and constriction of the ductus arteriosus. Circ Res 2013; 112(5): 802-15. [ Links ]
68. Zorzano A, Liesa M, Palacín M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int J Biochem Cell Biol 2009;41(10): 1846-54. [ Links ]
69. Sebastián D, Hernández-Alvarez MI, Segalés J, et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci U S A 2012;109(14):5523-8. [ Links ]
70. Zhang Z, Wakabayashi N, Wakabayashi J, et al. The dynaminrelated GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Mol Biol Cell 2011;22:2235-45. [ Links ]
71. Twig G, Elorza A, Molina AJ, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 2008;27(2):433-46. [ Links ]
72. Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest 2009;119(5):1275-85. [ Links ]
73. Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010;121:2012-22. [ Links ]
74. Sharp WW, Fang Y-H, Han M, et al. Dynamin related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemiareperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J 2014;28(1):316-26. [ Links ]
75. Chen Y, Liu Y, Dorn GW II. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res 2011;109(12):1327-31. [ Links ]
76. Papanicolaou KN, Kikuchi R, Ngoh GA, et al. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ Res 2012;111(8):1012-26. [ Links ]
77. Papanicolaou KN, Khairallah RJ, Ngoh GA, et al. Mitofusin-2 maintains mitochondrial structure and contributes to stressinduced permeability transition in cardiac myocytes. Mol Cell Biol 2011; 31(6):1309-28. [ Links ]
78. Lehninger A. Bioquímica: Las bases moleculares de la estructura y función celular. 4a edición. Barcelona. Ediciones Omega. 2006. Cap. 22:599-628. [ Links ]
79. De Berardinis R, Sayed N, Ditsworth D, et al. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev 2008;18(1):54-61. [ Links ]

