











 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
I. Introduction
Nuclear magnetic resonance (NMR) spectroscopy is a powerful technique for studying the structure of proteins under diferent conditions 1. This method is used for high-purity protein samples in an aqueous phase. In the case of spin-spin relaxation, the transverse component of the magnetization vector decays exponentially towards its equilibrium value in NMR. The time constant describing this process is known as T2. This name is based on T1, which is the time constant of the spin-lattice relaxation process. The short T2 time causes pulse loss during pulse sequences. On the other hand, with increasing protein size, this time is reduced. It is therefore difcult to prepare a solution for large proteins, so using this method for them is limited. By encapsulating the protein in the aqueous solution formed in the cavity of the reverse micelles, and then placing the complex in a non-polar solvent, this problem is largely resolved 2. In this case, the viscosity of the non-polar solvent must be much less than water 3. Short chain alkanes are a good candidate for this purpose 4. The nature of surfactants, the ability to dissolve surfactant in a non-polar solvent, and the ratio of water to surfactant are among the important parameters for the success of this method 5.
Flavodoxin is an electron transfer protein that plays a role in photosynthetic reactions 6. In avodoxins, a Flavin mono nucleotide molecule is attached to the protein in a non-covalent manner, which causes the redox property of proteins 7. From a structural standpoint, avdoxin is an protein in which the sheets are surrounded by helices 8. Since avodoxin is a simple onedomain protein, it is a suitable model for protein studies such as stability and folding. Cheung et al. have investigated the dynamics of avodoxin protein in crowded environments by molecular dynamics simulation 9. They showed that protein stability increases in in silico crowding conditions, and folding routes experiencing topological frustrations can be either relieved or enhanced upon manipulation of the crowding conditions. Studies have also shown that by removing the Flavin mononucleotide molecule from avodoxin and creating the Apoprotein form, the binding position does not change greatly; only the aromatic rings of tyrosine-94 and tryptophan-57 are closer to each other 6. Genzor et al. studied the stability of apo- avodoxin conformation in urea solution in diferent acidic and ionic strengths. They found that at a pH of between 6 and 11 apoavodoxin conformation is stable, whereas at a lower pH, between 2 and 3.5, proteins lose their stability 10. The structure of avodoxin in the presence of a cross-linked, spherical, sucrose-based polymer called Ficoll 70 has been studied experimentally and theoretically 11, 12. This circular dichroism and in silico study showed that the helical content of folded apo avodoxin was increased by additions of Ficoll 70. Other experimental evidence indicated that correct contact formation around the third -strand of avodoxin in the central sheet is crucial to continued folding to the native state in crowded media. Research has also shown that avodoxin in the presence of Dextran retains about 70% of its secondary structure 13.
In this manuscript, a simulation of molecular dynamics was used to study the avdoxin protein in a mixture of the surfactants 1-decanoyl-rac-glycerol and lauryldimethylamine-N-oxide. Pentane solvent was used for solute solvation. For comparison, molecular dynamics simulation of protein in water was also performed.
II. Computational details
For the molecular dynamics simulation, four simulation boxes were designed. In the rst box, the box dimensions were considered according to the size of the avodoxin, 5:95 5:95 5:95 nm3, and the protein was placed in the center of the box. Then the box was lled with water solvent (named as System A). In the second box, the protein was placed in the center of the box, with dimensions 6:75 6:75 6:75 nm3, then 130 1- decanoyl-rac-glycerol molecules and 70 molecules of lauryldimethylamine-N-oxide were randomly added to the box. Also, 50 molecules of water were added to this box, and nally, the box was lled with 1000 pentane molecules (named as System B). The specication of the third box was like the second box, only difering in that 5 hexanol molecules were also randomly placed in the box (named as System C). The fourth box was similar to the second box, but in this box, 10 hexanol molecules were randomly placed in it (named as System D). The components used in the various systems are summarized in Table 1. Since 1-decanoyl-rac-glycerol is non-ionic, hexanol solvent is used as auxiliary solvent. These molecular ratios are selected based on the reported optimal ratios in Dodevski et al.'s experimental work 14. The structure of avodoxin was obtained from the protein data bank with code 1obv 15. To perform molecular dynamics simulation, the Gromacs software version 5.2.1 was used 16. Charm27 all-atom force eld was used for calculations, since the force eld parameters for 1-decanoyl-rac-glycerol, lauryldimethylamine N-oxide, hexanol and pentane are not present in the default version of the Gromacs software. The structure of these compounds was optimized using the B3LYP density functional method with the basis function of 6-31G*. To control the optimization, frequency calculations were performed and virtual frequencies were not observed. The charge of each atom was calculated by the CHELPG method. All ab initio calculations were done by GAMESS software package 17. The optimized structure of the compounds is shown in Fig. 1.


The Swiss Param web server 18 was used to determine the force eld parameters of the studied compounds. Water model TIP3P 19 was used for water in simulation boxes. In order to neutralize the systems, an appropriate number of Na+ ions were added to each box. To x a constant temperature and pressure during the simulations, system components were coupled with V-rescale and Nose-Hoover thermostat 20 respectively, in each of the equilibration steps and molecular dynamics simulations. The temperature and pressure used in the calculations are 298 Kelvin and 1 bar, respectively. LINCS algorithm 21 was employed to x the chemical bonds between the atoms of the non-water molecules and SETTLE algorithm 22 was used in the case of water molecules. The PME algorithm was used to calculate electrostatic interactions 23. Energy minimization was done using the steepest descent method 24 to eliminate the primary kinetic energy and to eliminate inappropriate contacts between the atoms. Each simulation box achieved a two-stage equilibrium in NVT and NPT ensemble. At this stage, the time of equilibration was considered 10 ns with a time step of 2fs. Finally, molecular dynamics was performed by solving the second Newton equation for 140 ns with the 2fs time step. Each simulation was repeated four times under the diferent initial conditions, to avoid any dependency on the initial conditions and to increase the accuracy of the simulations.
III. Results and discussion
An important point when using surfactants in encapsulating proteins in NMR spectroscopy is to maintain the native protein structure and reduce its reorientation time. Therefore, the structural and dynamic parameters of avodoxin in the designed systems were calculated for analysis. One common method of displaying the stability of a protein's structure is to calculate the RMSD values during the simulation. The root mean square deviation (RMSD) is dened as:

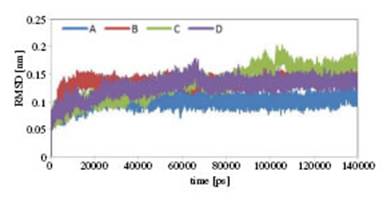


Figure 4: Average RMSF values for each residue of Flavodoxin during simulation in A, B, C and D systems.

According to the gure, protein stability in surfactant systems is observed to be somewhat reduced compared to system A. This decrease in stability for the C system is greater than for B and D. However, the difeerence in C-RMSD values in difierent systems is not high. On the other hand, given the fact that avodoxin has 169 residues, this difierence in stability in various systems is not significant. Another factor that can determine the activity and stability of the protein is compression of the conformation 25, in which the radius of gyration (Rg) indicates the compression of the protein structure. The Rg of a protein is calculated using the following equation:
Where mi is the atomic mass of i and ri the atomic position of i relative to the center of mass of the molecule 26. The Rg of the studied systems was calculated and the results are shown in Fig. 3. The Rg values of the proteins vary from 1.48 nm to 1.63 nm in difierent systems. Flavodoxin in system A has the smallest Rg and in system C the highest Rg. Since the compactness of the structure of the proteins is attributed to their activity 27, according to 3 the presence of avodoxin in the surfactant solution decreases its activity. The root mean square uctuation (RMSF) values of avodoxin sequences are calculated in difierent systems and shown in Fig. 4. Except for the residues valine-18, isoleucine-21, tryptophan-66 and glycine-68 in the avodoxin of system C, the exibility of other residues in system A is more than in the other systems. Another note- worthy point is that the C-terminal of the protein is less afected by the non-aqueous environment. In fact, the greatest difierence in the RMSF values in the various systems related to amino acids is 1 to 79. In this region, the helices and sheets in the secondary structure of avodoxin have smaller lengths than the helices and sheets in the c-terminal of the protein. If two faces are considered for proteins, the face of the N-terminal is more exposed to the environment. In avodoxins, the distance between two aromatic amino acids in the ligand binding site is considered as a measure of protein activity 8. The distance between the tryptophan 57 and phenylalanine-94 residues of avodoxin in system A is shown in Fig. 5.


According to the gure, this distance is 7.61 angstroms. The distance between these two amino acids in systems B, C and D was 9.48, 10.05 and 10.42 angstroms, respectively. According to the values obtained, avdoxin activity seems to decrease in surfactant solutions. Sampling of the suit- able structure for analysis in molecular dynamics trajectories has always been one of the interesting issues. Averaging the coordinates of the molecular coordinates in the last few nanoseconds of the simulation is one of the methods used in this regard 28. The sampling of coordinates at a time when the system is in equilibrium is another method used for this 29. In these methods, variations of a quantity such as C-RMSD in the area are used. However, there is no physical concept in the average of atomic coordinates 30, so for sampling of the simulationsthe free energy landscape (FEL) analysis method is used 31. There are three main stages of FEL analysis: calculation of the C-RMSD and the avodoxin radius of gyration (Rg), obtaining the possibility of the presence of avodoxin protein conformation in each corresponding value of C-RMSD and Rg, and calculation of the free energy of configurations based on the probability values of presence. The results of the FEL analysis are shown in 3D diagrams in Fig. 6 for A, B, C and D systems, respectively. In these gures, the minimum regions of free energy are shown in blue. According to these gures, it can be seen that all systems have a local minimum with the least amount of free energy. Regarding the values of C-RMSD and Rg corresponding to the lowest free energy, the structure of avodoxin was sampled from the molecular trajectories in the simulations. Then, using the software polyview 2D 32, the secondary structure of the avodoxin obtained in the previous step was analyzed. The result is shown in Fig. 7 for difierent systems. As the color darkens, it indicates that the level of solvent-accessible surface area for the residue decreases. With respect to the gures, it is observed that in the B system, the helices between aspartic-35 and aspartic-46 are eliminated. The beta sheet between the Valin-31 and Aspartic-35 sequences has also been shortened. The downsizing of this beta sheet in C and D systems is less than in the B system. In these areas, by changing the structure of the helices and sheets, the level of solvent-accessible surface area of residues has increased somewhat. The region of change for the C system is related to the helices between the tryptophan-57 and the aspartic-75 residues. In general, the secondary structure of the avodoxin in the c-terminal region has changed little. These results are consistent with the results of the RMSF values.
A protein contact map is a convenient way to determine changes in the tertiary structure of proteins in difierent conditions. The avodoxin contact map was obtained in various simulations and the results are shown in Fig. 8. In these gures, the avodoxin structure of system A was considered as a wild structure, and the remaining structures were compared to system A. In these gures, the lower half is the contact map of avodoxin, while the upper half is the protein difierence contact map; they are shown in difierent systems relative to system A.

Figure 7: The secondary structure of the Flavodoxin, along with the solvent-accessible surface area for its residues in systems A (top-left), B (top-right), C (bottom-left) and D (bottom-right).

Figure 8: (Left) The contact map of Flavodoxin in B (left-lower triangle) and difierent contact map of Flavodoxin in B and Flavodoxin in A (right-upper triangle). (Center) The contact map of Flavodoxin in C (left-lower triangle) and difierent contact map of Flavodoxin in C and Flavodoxin in A (right-upper triangle). (Right) The contact map of Flavodoxin in D (left-lower triangle) and difierent contact map of Flavodoxin in D and Flavodoxin in A (right-upper triangle). The colors used in the gure are described in the main text.
In the lower part of the gure, the black color represents the common contacts and the green color indicates the contacts that are in the wild structure of system A, but not in the structure of the secondary system; the red color represents the contacts that are in the secondary structure but not in the structure of system A. In the upper part, the blue color indicates the regions with the smallest difierences and the red color indicates the areas with the largest difierences. The greater the difierence in red color between Figs. 8 (center) and 8 (right) in comparison with Fig. 8 (left), the more the tertiary structure of avodoxin in systems C and D has been a ected. Also, areas of tertiary structure change in these systems are more focused than in system B. Finally, to determine the dynamics of avodoxin, its difiusion coeficient was calculated. Prediction of the difiusion coeficients of molecules in solvent is of theoretical importance. As the difiusion coeficient value increases, the mobility of the molecule increases 33. The Einstein relation was used to calculate the difiusion coeficient of avodoxin in all simulated systems:

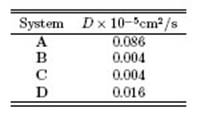
With regard to the difusion coeficient, the avodoxin dynamics are slower in systems B, C and D than in system A. Thus, the time of reorientation of protein conformation in systems B, C and D is less than that of system A. In other words, using this surfactant mixture can lead to a better NMR spectrum. This result is consistent with the results in the experimental work of Dodevski et al 14.
IV. Conclusions
By encapsulating the protein in the aqueous solution formed in the cavity of the reverse micelles, and then placing the complex in a non-polar solvent, the problem of slow tumbling will be greatly resolved in NMR spectroscopy. In this work, a mixture of the surfactants 1-decanoyl-rac-glycerol and lauryldimethylamine-N-oxide were used, together with the auxiliary solvent hexanol. Changes in the structure and dynamics of avodoxin protein were studied in this mixture. Pentane solvent was used as a low viscosity solvent. Compared with the structure of avodoxin in water, the simulation results indicate that the protein's secondary and tertiary structures in the mixture of surfactants are altered. However, its dynamics decrease. It was observed that in the absence of hexanol the helical and sheet content of the protein decreased in difierent regions. Also, in the presence of hexanol the ordered secondary structure of the protein decreased relative to two component systems consisting of protein and water. However, the level of reduction of the regular secondary structure was lower than in the absence of hexanol. Decreasing the areas of the regular secondary structure made the sequences more accessible to solvent. In general, the secondary structure of the avodoxin in the c-terminal region changed little. The difierent data obtained from the simulation are consistent with each other. The results are also in agreement with previous work in this area 35.

