










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkBAG. Journal of basic and applied genetics
versión On-line ISSN 1852-6233
BAG, J. basic appl. genet. vol.24 no.2 Ciudad Autónoma de Buenos Aires dic. 2013
ARTÍCULOS ORIGINALES
Mapeo de QTL para una medida multivariada de la reacción al virus del mal de Río cuarto
Bonamico N.C.1*, Di Renzo M.A.1, Borghi M.L.1, Ibañez M.A.1, Díaz D.G.2, Salerno J.C.2, Balzarini M.G.3
1 Facultad de Agronomía y Veterinaria, Universidad Nacional de Río Cuarto, Agencia N°3, 5800 Río Cuarto, Argentina
2 Instituto de Genética 'Ewald A. Favret', Instituto Nacional de Tecnología Agropecuaria, cc 25, 1712 Castelar, Argentina
3 Facultad de Ciencias Agrarias, Universidad Nacional de Córdoba y CONICET (Consejo Nacional de Investigaciones Científicas y Técnicas), cc 509, 5000 Córdoba, Argentina
nbonamico@ayv.unrc.edu.ar
RESUMEN
El índice de severidad de enfermedad (ISE), un indicador de resistencia multidimensional, es una media ponderada de la severidad e incidencia de la enfermedad del maíz causada por el Mal de Río Cuarto Virus (MRCV). En el presente estudio se realizó un mapeo por intervalo con el objetivo de identificar loci de caracteres cuantitativos o QTL ("quantitative trait loci") asociados con el ISE del Mal de Río Cuarto (MRC) en una población de líneas endocriadas recombinantes (RILs) de maíz evaluadas en ambientes donde la enfermedad es endémica. Los resultados sugieren que algunas regiones genómicas (cromosomas 1, 4, 6, 8 y 10) tienen un efecto significativo sobre la reacción al MRCV. Alrededor del 40% de los intervalos significativos coinciden con aquellos identificados en estudios previos. Por otro lado, se identificaron intervalos no detectados cuando el análisis se realizó carácter por carácter. Los resultados sugieren que es posible seleccionar genotipos resistentes al virus utilizando tanto los síntomas particulares de la enfermedad como QTL asociados al MRC.
Palabras clave: Maíz; Líneas endocriadas recombinantes; Índice de severidad de enfermedad; Microsatélites; Enfermedad viral.
ABSTRACT
The disease severity index (DSI), a multidimensional indicator of resistance, is a weighted average of the severity and incidence of the disease of maize caused by the Mal de Río Cuarto Virus (MRCV). Interval mapping was used in order to detect quantitative trait loci (QTL) associated with the DSI of the Mal de Río Cuarto (MRC) disease in a population of recombinant inbred lines (RILs) of maize tested in environments where the disease is endemic. The results suggest the existence of genomic regions (chromosomes 1, 4, 6, 8 and 10) with a significant effect on MRC. About 40% of the significant intervals were consistent with those identified in previous studies. However, other intervals were associated with the simultaneous expression of symptoms that are not detected when the analysis is performed trait by trait. The results suggest that virus-resistant genotypes can be selected using the particular symptoms of the disease as well as using MRC-QTL.
Key words: Maize; Recombinant inbred lines; Disease severity index; Microsatellites; Viral disease.
INTRODUCCIÓN
El Mal de Río Cuarto (MRC) es la patología más importante del maíz en Argentina (Redinbaugh y Pratt, 2009). En años de gran incidencia, se registraron lotes con pérdidas en los rendimientos superiores al 70% (Rodriguez Pardina et al., 1998). El agente causal del MRC, Mal de Río Cuarto virus o MRCV por sus siglas en inglés, pertenece al género Fijivirus familia Reoviridae (Distéfano et al., 2002). Este virus es naturalmente transmitido por medio del insecto vector Delphacodes kuscheli en forma persistente, circulativa y propagativa, por lo que el vector es reservorio natural y la población de macrópteros migrantes constituye el principal inóculo de la enfermedad (Ornaghi et al., 1993).
La ocurrencia y magnitud de las enfermedades virales en la naturaleza dependen de la interacción del vector con el patógeno, el hospedante y el ambiente. El control y la prevención de estas enfermedades provienen de la ruptura de alguna de las interacciones en el tetraedro de la enfermedad (Redinbaugh y Pratt, 2009).
La presencia de enaciones sobre las nervaduras en el envés de las hojas constituye el síntoma característico y distintivo del MRC. Debido a las modificaciones en los niveles hormonales endógenos se presentan otros síntomas como tallos achatados con entrenudos cortos, hojas del tercio superior recortadas o reducidas, panojas atrofiadas de escaso tamaño y espigas múltiples con pocos o sin granos (March et al., 1995). El modo más eficaz para controlar enfermedades virósicas en cultivos extensivos es la incorporación de resistencia genética (Di Renzo et al., 2002; Di Renzo et al., 2004; Redinbaugh y Pratt, 2009).
La biología molecular y la bioinformática ayudan a localizar loci de caracteres cuantitativos ("quantitative trait loci" o QTL) y permiten evaluar los efectos de genes individuales y las interacciones entre ellos y con el ambiente. La identificación de QTL para diferentes enfermedades y la consecuente selección indirecta favorecen la efectiva acumulación de genes de resistencia cuantitativa (Asíns, 2002; Lucas, 2010). La Organización de las Naciones Unidas para la Alimentación y la Agricultura (FAO) promueve la aplicación de la selección asistida por marcadores (marker-assisted selection o MAS), la cual es de uso frecuente en los actuales programas de mejoramiento genético de maíz tanto públicos como privados (Ragoty Lee, 2007).
Numerosos trabajos han identificado QTL para resistencia a virus mediante el uso de microsatélites (SSR) (Wu et al., 2007; Ingvardsen et al., 2010). La eficiencia para manipular QTL depende de su identificación y ubicación precisa así como de la consistencia en diferentes generaciones, fondos genéticos y ambientes (Rafalski, 2002). Trabajos realizados con una población de mapeo F2:3, de fondo genético similar al del presente estudio, permitieron estimar valores moderados de heredabilidad del carácter resistencia al MRC (Di Renzo et al., 2002) y realizar análisis de asociación que indicaron dos posibles QTLs en los grupos de ligamiento 1 y 8 (Di Renzo et al., 2004). Los resultados obtenidos con una población F2:3 de mapeo producto de un cruzamiento biparental diferente al utilizado por Di Renzo et al. (2004) permitieron identificar QTL relacionados a los síntomas de MRC en los cromosomas 1, 4, 8 y 10 (Kreff et al., 2006). En un trabajo previo (Bonamico et al., 2010), empleando un método de análisis multivariado, sugerimos la presencia de un grupo reducido de SSR asociados significativamente con diferentes síntomas del MRC. El mapeo de QTL para caracteres asociados a cada uno de los síntomas del MRC (Bonamico, 2010) y para los caracteres incidencia y severidad (Bonamico et al., 2012) permite inferir que el mapeo de un carácter sintético o matemáticamente construido a partir de la combinación de estas dimensiones de análisis constituye una alternativa para seleccionar genotipos resistentes al MRC. Dada la existencia de diversos síntomas que se manifiestan en grados diversos y con distinta incidencia en las poblaciones bajo selección, se ha propuesto el cálculo de índices de resistencia como el índice de severidad de enfermedad (ISE) (Grau et al., 1982). Este índice constituye una media de la severidad de la reacción al MRCV clasificada según la expresión simultánea de los síntomas característicos, ponderada por la incidencia de los distintos grados de severidad en un conjunto de plantas del genotipo (Di Renzo et al., 2004). La identificación de QTL asociados con el ISE y su validación con QTL previamente mapeados para resistencia al MRC en maíz es crucial para poder desarrollar estrategias efectivas de mejoramiento genético. Estudios de validación han revelado que hay pocos QTL comunes entre diferentes generaciones de poblaciones de mapeo y entre ambientes de evaluación en una misma generación (Melchinger et al., 1998).
Los objetivos de este trabajo fueron: i) mapear QTL asociados al ISE para la reacción al MRCV en una población de líneas endocriadas recombinantes de maíz; ii) analizar la consistencia entre las regiones genómicas aso ciadas al ISE y las identificadas con otras formas de estimar la reacción al MRCV.
MATERIALES Y MÉTODOS
Material vegetal
Una población de líneas endocriadas recombinantes (RILs) (F2:6) de maíz derivadas del cruzamiento entre la línea BLS14 (resistente) y la línea Mo17 (susceptible) se evaluó por su reacción a la infección del MRCV mediante caracteres fenotípicos relacionados con la expresión de la enfermedad. Estas RILs son descendientes de la población F2:3 utilizada en Di Renzo et al. (2004) para un estudio de mapeo de QTL para resistencia al MRC.
Ensayo de campo
Los ensayos de campo se realizaron en un diseño en bloques completos al azar (DBCA) con dos repeticiones, bajo condiciones de transmisión natural en dos localidades de la zona sur de la provincia de Córdoba: Río Cuarto (33º 8'S, 64º 20'O, 334 msnm, 630 mm de precipitación anual) y Sampacho (33º 19'S, 64º 42'O; 510 msnm, 676 mm de precipitación anual). Los ensayos se establecieron en los ciclos agrícolas 2004/05, 2005/06 y 2006/07. Cada una de las seis combinaciones localidad-año se consideró un ambiente: Río Cuarto 2005 (R5), Río Cuarto 2006 (R6), Río Cuarto 2007 (R7), Sampacho 2005 (S5), Sampacho 2006 (S6) y Sampacho 2007 (S7). Las líneas parentales se incluyeron en cada ambiente. El número de plantas osciló entre 10 y 15 espaciadas a 20 cm sobre el surco. Las labores de presiembra fueron las convencionales. Las malezas preemergentes y postemergentes fueron controladas con agroquímicos y en forma manual, respectivamente.
Evaluación fenotípica
La reacción a la infección del MRCV de cada una de las RILs se estimó mediante el ISE. Para calcular este índice, las plantas de cada genotipo fueron clasificadas según el grado de severidad de enfermedad propuesto por Ornaghi et al. (1999), expresado en una escala que consta de cuatro grados (0, 1, 2 y 3) según la simultaneidad de síntomas observados. El grado 0 corresponde a una planta asintomática; el grado 1 a una planta que presenta enaciones, con o sin disminución de altura; el grado 2 a una planta con presencia de enaciones, reducción del tamaño de la panoja, láminas foliares atrofiadas en el tercio superior, y espigas generalmente curvas y pequeñas; y el grado 3 a una planta con síntomas muy severos, enaciones, acortamiento de entrenudos, producción de granos por espiga escasa o nula, y espigas pequeñas, múltiples y sin granos. Grau et al. (1982) propusieron la siguiente ecuación para el cálculo del ISE, que puede tomar valores de 0 a 100, siendo este último valor el correspondiente a genotipos severamente afectados:

Evaluación genotípica
El ADN de las RILs se extrajo a partir de tejido foliar liofilizado de plantas de maíz jóvenes crecidas en invernáculo, según el método utilizado en Di Renzo et al. (2004). Un conjunto de 150 marcadores moleculares SSR se evaluó en las líneas parentales BLS14 y Mo17. Del total de marcadores, 113 revelaron bandas nítidas y de éstos sólo los polimórficos se utilizaron para analizar la población de RILs. Para la construcción del mapa de ligamiento, se eligieron los marcadores polimórficos con el menor número de datos faltantes. Los perfiles de ADN obtenidos para cada RIL correspondieron a marcadores SSR pertenecientes a ocho cromosomas de maíz (1, 2, 3, 4, 6, 8, 9 y 10). El 50% de los SSR probados pertenecen a los grupos de ligamiento 1, 4, 8 y 10, en los que se detectaron QTL para resistencia al MRC (Di Renzo et al., 2004; Kreff et al., 2006). Las secuencias de los marcadores SSR se obtuvieron a partir de la base de datos genéticos de maíz (MaizeGDB, http://www.maizegdb.org) y se sintetizaron en el laboratorio Alpha DNA (http://www.alphadna.com). La amplificación por PCR se realizó según el protocolo utilizado en Di Renzo et al. (2004).
Mapeo de QTL
El mapeo de los QTL se realizó con el programa Plabqtl versión 1.0 (Utz y Melchinger, 1995) usando un mapa genético construido a partir de la misma población de RILs (Bonamico et al., 2012). Los análisis se efectuaron por ambiente y a partir del ensayo multiambiental, usando los datos fenotípicos de cada ambiente, se ajustó primero un modelo lineal mixto (MLM) (Littell et al., 2006) para obtener los estimadores del ISE de cada línea, corregidos por posibles correlaciones experimentales. Este modelo incluyó el efecto de bloque como fijo y una matriz de varianza y covarianza residual con estructura espacial de tipo exponencial para contemplar la posible auto-correlación entre parcelas dentro de cada ambiente. No obstante, la comparación de este modelo con el modelo de independencia sugirió que las medias aritméticas de cada genotipo en cada ambiente podían ser usadas en los análisis subsecuentes. Con estas medias se emplearon los métodos de mapeo por intervalo simple (SIM, Modelo III) y por intervalo compuesto (CIM, Modelo II) (Zeng, 1994). En el CIM se utilizaron marcadores moleculares como cofactores. El logaritmo de las probabilidades (log of the odds o LOD) mínimo para declarar la presencia de un posible QTL fue 2.5. La posición del QTL fue asignada al punto del intervalo de análisis donde el valor de LOD fue máximo. El efecto genético de aditividad fue estimado como la mitad de la diferencia entre el valor de los dos homocigotas, asumiendo que el segundo parental es quien aporta alelos favorables para la variable en estudio (Falconer, 1989). Valores negativos indican que el parental BLS14 aporta alelos favorables para disminuir el ISE. La variación fenotípica total del ISE, explicada por los posibles QTL detectados, se estimó a través de un modelo de regresión múltiple.
RESULTADOS
Evaluación fenotípica
El ISE de las RILs mostró una distribución de frecuencias próxima a la normal al considerar el conjunto de ambientes de evaluación (Figura 1). El valor medio en las RILs fue aproximadamente intermedio al de los parentales. El valor medio del ISE en el parental resistente fue menor al 10%, mientras que el parental susceptible presentó un valor superior al 80% del máximo de la escala.
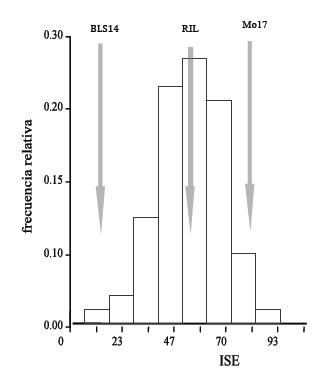
Figura 1. Frecuencia de distribución relativa del índice de severidad de enfermedad (ISE) del Mal de Río Cuarto en una población de líneas endocriadas recombinantes derivadas del cruzamiento entre las líneas BLS14 (resistente)× Mo17 (susceptible), evaluada en seis ambientes en donde la enfermedad es endémica.
La Tabla 1 muestra los valores del ISE del MRC obtenidos en las líneas BLS14 (resistente) y Mo17 (susceptible) y las RILs derivadas (F2:6), en seis ambientes de evaluación. El ensayo sembrado en la localidad de Río Cuarto en la campaña 2006/07 (R7) bajo condiciones de transmisión natural presentó infecciones severas, mientras que los ensayos realizados en la misma localidad en las campañas 2004/05 (R5) y 2005/06 (R6) escaparon al pico poblacional de los vectores de la enfermedad. Como resultado, en estos últimos dos ambientes, los materiales susceptibles no manifestaron síntomas tan severos. Los materiales evaluados en Sampacho durante los ciclos agrícolas 2004/05 (S5), 2005/06 (S6) y 2006/07 (S7) presentaron un adecuado nivel de síntomas. En el ciclo agrícola 2006/07 se observaron lotes en la zona en donde la enfermedad es endémica con un 100% de plantas afectadas por el MRCV. La Secretaría de Agricultura de la provincia de Córdoba estimó pérdidas económicas por MRC durante dicha campaña de 70 millones de dólares (Mattio et al., 2008).
Tabla 1. Valores del índice de severidad de enfermedad (ISE) del Mal de Río Cuarto obtenidos en las líneas BLS14 (resistente) y Mo17 (susceptible) y en las líneas endocriadas recombinantes derivadas (F2:6). Observaciones en seis ambientes en donde la enfermedad es endémica.
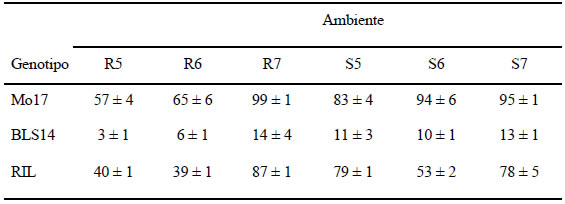
R5: Río Cuarto 2005; R6: Río Cuarto 2006; R7: Río Cuarto 2007; S5: Sampacho 2005; S6: Sampacho 2006 y S7: Sampacho 2007.
En general se observaron correlaciones positivas y significativas entre el ISE y distintos síntomas del MRC, lo cual indica que a mayor valor de afección en cada uno de los síntomas, mayores los valores del ISE (Tabla 2). Los valores de correlación más elevados con el ISE, en los diferentes ambientes, fueron estimados para presencia de enaciones y acortamiento de entrenudos. Estos síntomas serían los recomendables para la selección ambiente-específica de genotipos de maíz resistentes al MRC cuando no se calcule el ISE.
Tabla 2. Coeficientes de correlación entre el índice de severidad de enfermedad (ISE) y caracteres asociados a síntomas del Mal de Río Cuarto en seis ambientes en donde la enfermedad es endémica.
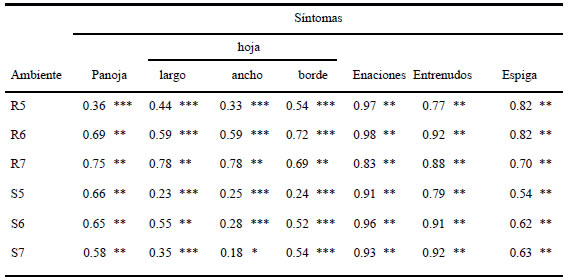
R5: Río Cuarto 2005; R6: Río Cuarto 2006; R7: Río Cuarto 2007; S5: Sampacho 2005; S6: Sampacho 2006 y S7: Sampacho 2007. **p<0.01; ***p<0.001.
Mapeo de QTL
Siete QTL fueron detectados para el ISE. En Sampacho 2005 se identificaron tres QTL y en Sampacho 2007 cuatro. En los ambientes Río Cuarto 2005, Río Cuarto 2007 y Sampacho 2006 se identificaron dos QTL significativos, mientras que en Río Cuarto 2006 se identificó sólo un QTL (Tabla 3).
Tabla 3. Localización y efectos de QTL para el índice de severidad de enfermedad (ISE) del Mal de Río Cuarto en ambientes individuales y a través de ambientes, en donde la enfermedad es endémica.
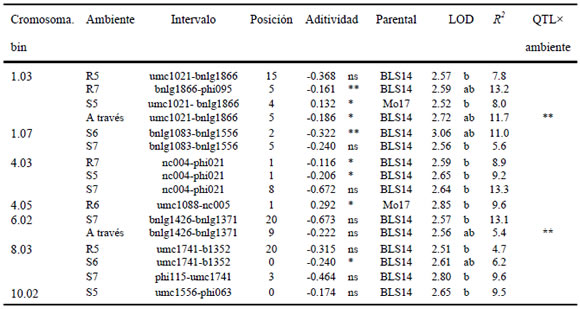
Ambiente: R5: Río Cuarto 2005; R6: Río Cuarto 2006; R7: Río Cuarto 2007; S5: Sampacho 2005; S6: Sampacho 2006 y S7: Sampacho 2007. Intervalo: referido al mapa publicado en Bonamico et al. (2012). Posición: distancia del QTL (cM) al marcador más próximo. Aditividad: efecto estimado como la mitad de la diferencia entre el valor de los dos homocigotas. Un valor negativo indica que el parental BLS14 aporta alelos para disminuir el valor del ISE. Nivel de significación, ns: no significativo; *<0.05; **<0.01. Letras a y b que acompañan al valor de LOD indican mapeo por intervalo simple (SIM) y compuesto (CIM), respectivamente. R²: proporción (%) de la variación fenotípica explicada.
Los siete QTL identificados para el ISE pertenecen a los cromosomas 1, 4, 6, 8 y 10. En los cromosomas 1 y 4 se identificaron dos QTL, mientras que en los cromosomas 6, 8 y 10 se identificó sólo un QTL en cada uno. De los siete QTL detectados, que se localizaron en los cromosomas 1 (bin 1.03 y 1.07), 4 (bin 4.03 y 4.05), 6 (bin 6.02), 8 (bin 8.03) y 10 (bin 10.02), dos fueron identificados de manera específica en los ambientes R6 y S5 en los cromosomas 4 (bin 4.05) y 10 (10.02), respectivamente.
En los ambientes R7, S5 y S7 prevaleció un mayor valor de afección de enfermedad reflejado en un mayor valor medio del ISE en las RILs. En relación a los marcadores SSR ligados a QTL para resistencia al MRC se observaron regiones del genoma asociadas con la resistencia al MRC de manera exclusiva con los ambientes de mayor y menor valor del ISE, respectivamente. En el grupo de ambientes de mayor ISE, se identificó en forma exclusiva un QTL en el cromosoma 4 (bin 4.03). En el grupo de ambientes de menor ISE, se identificó de manera exclusiva un QTL en el cromosoma 4 (bin 4.05). Por lo mencionado, se infiere que existen factores genéticos diferenciales en la regulación del carácter resistencia al MRC frente a diferentes niveles de ISE.
El porcentaje de variación fenotípica explicada en forma individual por cada QTL, tanto en ambientes individuales como a través de ambientes, fue de entre 5 y 13%. La variación fenotípica total explicada en forma conjunta por todos los posibles QTL detectados en cada uno de los ambientes individuales varió entre 5 y 18%. Uno de los QTL con mayor efecto sobre el ISE se identificó en el ambiente R7 en el cromosoma 1 (bin 1.03). Este QTL fue significativo de manera consistente en la mayoría de los ambientes de evaluación. Otros dos QTL en el ambiente S7, ubicados en los bins 4.03 y 6.02, explicaron en forma individual un máximo del 13% de la variación fenotípica del ISE.
Dado que se utilizaron RILs, sólo se estimaron los efectos genéticos de aditividad y de epistasis correspondiente, no siendo estos últimos estadísticamente significativos. La estimación de los efectos genéticos no evaluados podría ser de importancia menor ya que el híbrido entre las dos líneas parentales se comportó con una resistencia intermedia entre las mismas (datos no mostrados), lo que sugiere acción génica de tipo aditiva para el carácter.
Los QTL identificados como significativamente asociados con el ISE presentaron inconsistencia en los ambientes evaluados. No obstante, las regiones de los cromosomas 1, 4 y 8 (bins 1.03, 4.03 y 8.03) fueron consistentes en el 50% de los ambientes, incluso el del bin 1.03 en el análisis a través de ambientes. Los QTL identificados en el análisis a través de ambientes, pertenecientes a los cromosomas 1 y 6 (bins 1.03 y 6.02), presentaron interacción QTL×ambiente estadísticamente significativa (p= 0.01), es decir que no fueron detectados en todos los ambientes evaluados.
Los resultados del presente trabajo permitieron identificar regiones con efecto significativo sobre la reacción a la enfermedad MRC similares o cercanas a las informadas en trabajos anteriores (Di Renzo et al. 2004; Bonamico, 2010; Bonamico et al., 2010, 2012) (Tabla 4).
Tabla 4. Identificación de QTL y marcadores moleculares SSR asociados con la reacción al virus Mal de Río Cuarto.

Parental: S = Susceptible y R = Resistente.
Carácter: ISE = índice de severidad de enfermedad; INC = incidencia de enfermedad; SEV = severidad de enfermedad.
Método: SIM = Mapeo por intervalo simple; CIM = Mapeo por intervalo compuesto; ANAVA = Análisis de la varianza;
AD = Análisis discriminante discreto.
DISCUSIÓN
Evaluación fenotípica
La ocurrencia no sistemática de valores extremos de incidencia y severidad del MRC es común a través de los años. Severas epidemias de esta enfermedad ocurren cuando coinciden la presencia de genotipos susceptibles en estadios sensibles, un abundante reservorio del virus y altas densidades del insecto vector (Harpaz, 1972). La expresión de síntomas severos se relaciona con el estado fenológico del maíz al momento en que se produce la transmisión del virus y la máxima expresión de la enfermedad se da cuando genotipos susceptibles son infectados en estado de coleoptile a 3-4 hojas (March et al., 1995). Acorde a los rangos de valores, la segregación para el carácter fue de tipo transgresiva, es decir que algunas líneas se comportaron más susceptibles que Mo17 y otras más resistentes que BLS14. En trabajos realizados con generaciones tempranas F2:3 provenientes del mismo cruzamiento biparental se observó igual tipo de segregación (Di Renzo et al., 2002).
Mapeo de QTL
En maíz fueron identificados numerosos genes y/o QTL para resistencia a patógenos como hongos, bacterias y virus. Los cromosomas 1 y 4 de maíz contienen el mayor número de genes y/o QTL para resistencia a enfermedades causadas por estos patógenos. En los demás cromosomas también se identificaron genes y/o QTL para resistencia aunque en menor cantidad (Wang et al., 2007).
En estudios de resistencia a enfermedades, es frecuente la identificación de QTL ambiente-específicos, es decir que no son detectados en todos los ambientes de evaluación, lo cual puede ser valioso para la mejora genética si se consideran características ambientales particulares (Paterson et al., 1991).
Estudios previos de herencia y mapeo de QTL para resistencia al MRC han indicado que la expresión del carácter está determinada por efectos genéticos tanto aditivos como no aditivos (Presello et al., 1995; Di Renzo et al., 2004). El efecto genético aditivo de cada QTL indicó que los alelos favorables fueron aportados por ambas líneas parentales, tal como se reportó en Di Renzo et al. (2004). El parental tolerante al MRC aportó alelos que disminuyeron el ISE en seis de los siete posibles QTL identificados. Estos QTL correspondieron a los cromosomas 1, 4, 6, 8 y 10 (bins 1.03, 1.07, 4.03, 6.02, 8.03 y 10.02). En el bin 1.03, el parental resistente aportó alelos favorables cuando el marcador SSR bnlg1866 se mostró ligado de manera estadísticamente significativa al QTL.
Al comparar los QTL identificados para el ISE con los identificados en estudios previos para cada uno de los otros caracteres relacionados a síntomas del MRC, pueden observarse coincidencias tanto en posiciones como en la proporción de la variación fenotípica explicada. Se ha sugerido que caracteres altamente correlacionados tienen más posibilidades de compartir QTL (Lisec et al., 2008). En un trabajo previo (Bonamico, 2010), reportamos dos QTL para la presencia de enaciones y acortamiento de entrenudos, ubicados en los cromosomas 1 (bin 1.03) y 6 (bin 6.02), de los siete detectados para el ISE. Es decir que alrededor del 40% de los loci significativamente asociados con la reacción al MRCV fueron compartidos cuando se identificaron QTL para síntomas individuales o para el índice multidimensional. Estos QTL, detectados en la mitad de los ambientes evaluados, permitieron estimar el ISE y explicaron un máximo del 13% de la variación fenotípica observada. Por otra parte, se detectaron intervalos significativamente asociados con el ISE que no están relacionados con ninguno de los síntomas por sí solos (bins 1.07, 4.03, 4.05, 8.03 y 10.02). En el conjunto de los ambientes, los QTL comunes para el ISE y para síntomas individuales (Bonamico, 2010) se presentaron con correlaciones fenotípicas menores. Esto podría deberse a la existencia de un control genético diferencial o a una alta influencia de factores ambientales. Dado que se identificaron al menos dos QTL comunes entre el ISE, la presencia de enaciones y el acortamiento de entrenudos, una estrategia simple que podría resultar de interés a los mejoradores para seleccionar resistencia al MRC es usar estas variables fenotípicas de fácil y rápida evaluación. No obstante, el ISE es la variable más recomendable para mapear QTL para reacción a la infección del MRCV, debido a que en éste se considera tanto la severidad como la incidencia de la enfermedad.
Un aspecto importante en el uso de QTL para realizar selección indirecta es la consistencia de las posiciones de los QTL observadas en diferentes poblaciones de mapeo (Rafalski, 2002). En un trabajo previo (Di Renzo et al., 2004) donde se evaluó, mediante ISE, una generación temprana con el mismo fondo genético que la población utilizada en este trabajo, se identificaron dos QTL con efectos significativos sobre resistencia al MRC en los cromosomas 1 y 8 (bins 1.03 y 8.03/4). Posteriormente, al mapear el grado medio de severidad y otros caracteres relacionados a síntomas del MRC, se identificaron doce posibles QTL en los cromosomas 1 (bins 1.01, 1.03, 1.04, 1.06), 4 (bins 4.03, 4.05, 4.08), 6 (bins 6.00/2, 6.05), 8 (bins 8.03, 8.08) y 10 (bin 10.02) mediante el análisis de mapeo por intervalo, utilizando una población de RILs de maíz (Bonamico, 2010). También se identificó un grupo reducido de marcadores SSR significativos en los cromosomas 1 (bins 1.02, 1.06), 2 (bin 2.06), 6 (bin 6.02) y 8 (bin 8.08) mediante un análisis multivariado (Bonamico et al., 2010). El mapeo de QTL para los caracteres incidencia y severidad permitió identificar cuatro QTL localizados en los cromosomas 1, 4 y 10 (Bonamico et al., 2012). Al comparar los resultados con los informados por Kreff et al. (2006), quienes trabajaron con una población de mapeo de fondo genético distinto al utilizado en el presente estudio, se observa consistencia en varios de los QTL detectados (Tabla 4). Estos autores informaron que los QTL para resistencia al MRC están ubicados en los cromosomas 1, 4, 8 y 10. No obstante, se encontraron variaciones respecto a los resultados obtenidos por Kreff et al. (2006), ya que para el carácter ISE se identificaron dos nuevos QTL no informados por estos autores, en los bins 6.02 y 10.02. La similitud de los resultados obtenidos con los de trabajos previos no es una característica común ya que la acción de diversos factores como el uso de diferentes parentales, el tipo y el tamaño de la población de mapeo, la metodología empleada en la detección de QTL, y las diferentes condiciones climáticas y de cultivo usualmente atentan contra la consistencia de los resultados (Nair et al., 2005).
McMullen y Simcox (1995) sugirieron que, en el genoma de maíz, los genes de resistencia a diferentes patógenos frecuentemente se ubican agrupados e indican regiones genómicas involucradas en la expresión de resistencia a virus en los cromosomas 1 y 6. Otras revisiones recientes en maíz también sugieren que los genes y loci que confieren resistencia a diferentes virus no están distribuidos al azar en el genoma. Wisser et al. (2006) indicaron la presencia de dos grupos tanto en el cromosoma 1 como en el cromosoma 3, y un grupo en cada uno de los cromosomas 2, 4, 5 y 10. Redinbaugh y Pratt (2009) mencionaron cuatro grupos en los cromosomas 1, 3, 6 y 10. El presente trabajo sugiere que los QTL asociados con la reacción al MRCV, cuantificada a través del ISE, están en los cromosomas 1, 4, 6, 8 y 10. Estos resultados favorecen al desarrollo de conocimientos que son requeridos para la implementación de la selección indirecta mediada por marcadores (MAS) en el mejoramiento de la resistencia a la virosis MRC en maíz.
Alrededor del 43% de los intervalos detectados como significativos fueron los mismos que aquellos identificados en estudios previos que usaron cada unos de los síntomas como caracteres de mapeo (Bonamico, 2010). No obstante, surgieron algunos otros intervalos que estuvieron asociados con la expresión simultánea de los síntomas y que no se detectaron al realizar el análisis carácter por carácter. Por ello, la opción que resulta recomendable es la selección de genotipos resistentes vía marcadores asociados al ISE del MRC.
AGRADECIMIENTOS
Este trabajo fue subsidiado por la Agencia Nacional de Promoción Científica y Tecnológica (PICT N° 02231/07 BID), por la Agencia Córdoba Ciencia (PID N° 38) y por la Secretaría de Ciencia y Tecnología de la Universidad Nacional de Río Cuarto (PPI 2012-14), Argentina.
BIBLIOGRAFÍA
1. Asíns M.J. (2002) Present and future of quantitative trait locus analysis in plant breeding. Plant Breeding 121: 281-291. [ Links ]
2. Bonamico N. (2010) Mapeo de QTL en líneas endocriadas recombinantes de maíz: Regiones del genoma que confieren tolerancia a Mal de Río Cuarto. Tesis Doctoral. Universidad Nacional de Río Cuarto, Río Cuarto, Córdoba, Argentina. [ Links ]
3. Bonamico N.C., Balzarini M.G., Arroyo A.T., Ibañez M.A., Díaz D.G., Salerno J.C., Di Renzo M.A. (2010) Association between microsatellites and resistance to Mal de Río Cuarto in maize by discriminant analysis. FYTON 79: 31-38. [ Links ]
4. Bonamico N.C., Di Renzo M.A., Ibañez M.A., Borghi M.L., Díaz D.G., Salerno J.C. (2012) QTL analysis of resistance to Mal de Río Cuarto disease in maize using a RIL population. J. Agr. Sci. 150 (5): 619-629. (CJO doi: 10.1017/ S0021859611000943). [ Links ]
5. Di Renzo M.A., Bonamico N.C., Díaz D.D., Salerno J.C., Ibañez M.A., Gesumaría J.J. (2002) Inheritance of resistance to Mal de Río Cuarto (MRC) disease in Zea mays (L.). J. Agr. Sci. 139: 47-53. [ Links ]
6. Di Renzo M.A., Bonamico N.C., Díaz D.D., Ibañez M.A., Faricelli M.E., Balzarini M.G., Salerno J.C. (2004) Microsatellite markers linked to QTL for resistance to Mal de Río Cuarto disease in Zea mays L. J. Agr. Sci. 142: 289-295. [ Links ]
7. Distéfano A.J., Conci L.R., Muñoz Hidalgo M., Guzmán F.A., Hopp H.E., Del Vas M. (2002) Sequence analysis of genome segments S4 and S8 of Mal de Río Cuarto virus (MRCV): evidence that the virus should be a separate Fijivirus species. Arch. Virol. 147: 1699-1709. [ Links ]
8. Falconer D.S. (1986) Introduction to Quantitative Genetics. Longman Scientific & Technical, Essex, England. [ Links ]
9. Grau C.R., Radke V.L., Gillespie F.L. (1982) Resistance of soybean cultivars to Sclerotinia sclerotiorum. Plant Dis. 66: 506-508. [ Links ]
10. Harpaz I. (1972) Maize rough dwarf. A planthopper virus disease affecting maize, rice, small grains and grasses. Israel University Press, Jerusalem. p. 251. [ Links ]
11. Ingvardsen C.R., Xing Y., Frei U.K., Lübberstedt T. (2010) Genetic and physical fine mapping of Scmv2, a potyvirus resistance gene in maize. Theor. Appl. Genet. 120: 1621-1634. [ Links ]
12. Kreff E.D., Pacheco M.G., Díaz D.G., Robredo C.G., Puecher D., Céliz A., Salerno J.C. (2006) Resistance to Mal de Río Cuarto Virus in maize: A QTL Analysis. JBAG 17: 41-50. [ Links ]
13. Lisec J., Meyer R., Steinfath M., Redestig H., Becher M., Witucka-Wall H., Fiehn O., Törjék O., Selbig J., Altmann T., Willmitzer L. 2008. Identification of metabolic and biomass QTL in Arabidopsis thaliana in a parallel analysis of RIL and IL populations. Plant J. 53: 960-972. [ Links ]
14. Littell R.C., Milliken G.A., Stroup W.W., Wolfinger R.D., Schabenberger O. (2006) SAS for Mixed Models. Second Edition, Cary, NC: SAS Institute Inc. [ Links ]
15. Lucas J.A. (2010) Advances in plant disease and pest management. J. Agr. Sci. 149: 91-114. [ Links ]
16. March G.J., Balzarini M., Ornaghi J.A., Beviacqua J.E., Marinelli A. (1995) Predictive model for "Mal de Río Cuarto" disease intensity. Plant Dis. 79: 1051-1053. [ Links ]
17. Mattio M.F., Cassol A., Remes Lenicov A.M., Truol G. (2008) Tagosodes orizicolus: nuevo vector potencial del Mal de Río Cuarto. Trop. Plant Pathol. 33: 237-240. [ Links ]
18. McMullen M.D., Simcox K.D. (1995) Genomic organization of disease and insect resistance genes in maize. Mol. Plant Microbe. In. 8: 811-815. [ Links ]
19. Melchinger A.E., Utz H.F., Schön C.C. (1998) Quantitative trait locus (QTL) mapping using different testers and independent population sample in maize reveals low power of QTL detection and large bias in estimates of QTL effects. Genetics 149: 383-403. [ Links ]
20. Nair S.K., Prasanna B.M., Garg A., Rathore R.S., Setty T.A.S., Singh N.N. (2005) Identification and validation of QTLs conferring resistance to sorghum downy mildew (Peronosclerospora sorghi) and Rajasthan downy mildew (P. heteropogoni) in maize. Theor. Appl. Genet. 110: 1384-1392. [ Links ]
21. Ornaghi J.A., Boito G., Sánchez G., March G., Beviacqua J. (1993) Studies on the populations of Delphacodes kuscheli Fennah in different years and agricultural areas. J. Genet. & Breed. 47: 277-282. [ Links ]
22. Ornaghi J.A., March G.J., Boito G.T., Marinelli A., Beviacqua J.E., Giuggia J., Lenardón S.L. (1999) Infectivity in natural populations of Delphacodes kuscheli vector of "Mal Río Cuarto" Virus. Maydica 44: 219-223. [ Links ]
23. Paterson A.H., Damon S., Hewitt J.D., Zamir D., Rabinowitch H.D., Lincoln S.E., Lander E.S., Tanksley S.D. (1991) Mendelian factors underlying quantitative traits in tomato: comparison across species, generations, and environments. Genetics 127: 181-197. [ Links ]
24. Presello D., Céliz A., Frutos E. (1995) Efectos genéticos asociados con la resistencia a la enfermedad Mal de Río Cuarto en líneas endocriadas de maíz. En Avila L.G., Céspedes L.M. (Eds) Proceedings of III Latin American and XVI Andean Zone of Maize Researchers Meeting. Tomo I. Fundación Patiño SI. Bolivia. pp. 407-413. [ Links ]
25. Rafalski J.A. (2002) Plant genomics: present state and a perspective on future developments. Brief. Funct. Genomic Proteomic. 1: 80-94. [ Links ]
26. Ragot M., Lee M. (2007) Marker-assisted selection in maize: current status, potential, limitations and perspectives from the private and public sectors. En: Guimaraes E., Ruane J., Scherf B., Sonnino A., Dargie J. (Eds) Marker-Assisted Selection. Current status and future perspectives in crops, livestock, forestry and fish. FAO. pp 117-150. [ Links ]
27. Redinbaugh M.G., Pratt R.C. (2009) Virus Resistance. En: Bennetzen J.L., Hake S.C. (Eds) Handbook of Maize: Its Biology. pp 251-270. [ Links ]
28. Rodriguez Pardina P.E., Giménez Pecci M.P., Laguna I.G., Dagoberto E., Truol G. (1998) Wheat: A new natural host for the Mal de Río Cuarto virus in the endemic disease area, Río Cuarto, Córdoba Province, Argentina. Plant Dis. 82: 149-152. [ Links ]
29. Utz H.F., Melchinger A.E. (1995) PLABQTL. A computer program to map QTL. Institute of Plant Breeding, Seed Science, and Population Genetics. University of Hohenheim, D-70593 Stuttgart, Germany. [ Links ]
30. Wang G.X., Chen Y., Zhao J.R., Li L., Korban S.S., Wang F.G., Li J.S., Dai J.R., Xu M.L. (2007) Mapping of Defense Response Gene Homologs and Their Association with Resistance Loci in Maize. Journal of Integrative Plant Biology 49 (11): 1580-1598. [ Links ]
31. Wisser R.J., Balint-Kurti P.J., Nelson R.J. (2006) The genetic architecture of disease resistance in maize: a synthesis of published studies. Mini-Review. Phytopathology 96: 120-129. [ Links ]
32. Wu J., Ding J., Du Y., Xu Y., Zhang X. (2007) Genetic analysis and molecular mapping of two dominant complementary genes determining resistance to sugarcane mosaic virus in maize. Euphytica 156: 355-364. [ Links ]
33. Zeng Z.B. (1994) Precision mapping of Quantitative Trait Loci. Genetics 136: 1457-1468. [ Links ]

