










Services on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkBAG. Journal of basic and applied genetics
On-line version ISSN 1852-6233
BAG, J. basic appl. genet. vol.24 no.2 Ciudad Autónoma de Buenos Aires Dec. 2013
ARTÍCULOS ORIGINALES
Evaluation of six nucleotide polymorphisms for bovine traceability in the context of the Argentine-Chinese beef trade
Ripoli M.V.1, Wei S.2, Rogberg-Muñoz A.1, Guo B.L.2, Goszczynski D.E.1, Fernandez M.E.1, Mellucci L. 3, Lirón J.P.1, Villarreal E. 3, Wei Y.M.2, Giovambattista G1.
1 Instituto de Genética Veterinaria (IGEVET), CCT La Plata - CONICET - Facultad de Ciencias Veterinarias, Universidad Nacional de La Plata, La Plata, Argentina.
2 Key Laboratory of Agro-Products Processing and Quality Control, Ministry of Agriculture, Institute of Agro-Products Processing Science and Technology, Chinese Academy of Agricultural Sciences, P.O. Box 5109, Beijing, P.R. of China, 100193.
3 Unidad Integrada Balcarce (UIB), Facultad de Ciencias Agrarias, UNMDP-EEA (INTA) Balcarce.
Corresponding author: G Giovambattista. IGEVET - CCT La Plata - FCV - CONICET, Facultad de Ciencias Veterinarias, Universidad Nacional de La Plata, La Plata B1900AVW, CC 296, Argentina. Phone/fax 54-221-4211799. ggiovam@fcv.unlp.edu.ar
ABSTRACT
Genetic traceability refers to methods associated with the identification of animals and their products through DNA characterization of individuals, breeds or species. To trace breeds, it is necessary to define the breed groups to analyze, and the most appropriate molecular marker set. The selection of genetic markers depends on the gene frequency distribution, the genetic distance among breeds and the presence of private alleles. In this study, we assessed six single nucleotide polymorphisms (SNPs) located in the DGAT1, TG, LEP, GH, FABP4 and GnRHR genes, as potential genetic markers to be included into a panel for genetic traceability for the identification of breed origin associated with the bovine beef trade. The results of the genetic characterization of four of the main Chinese cattle populations and of the principal breeds raised in Argentina and in the world (five Bos taurus and two B. indicus) suggest that these SNP markers can be successfully used as a part of an effective traceability system for the identification of cattle breed origin in the context of the Chinese meat imports, and in particular in the Argentine-Chinese beef trade.
Key words: Breed traceability; Genetic traceability; Single nucleotide polymorphism; Chinese cattle; Argentine cattle.
RESUMEN
La trazabilidad genética, la cual se basa en la identificación de animales y sus productos, permite la identificación individual, racial o de especie. Esta metodología es útil para detectar fraudes y valorizar producciones locales. Para llevar a cabo la trazabilidad es necesario definir los grupos raciales a analizar y el panel de marcadores más apropiados a utilizar. La selección de marcadores depende de la distribución de las frecuencias génicas, de la distancia genética entre las razas y de la presencia de alelos privativos. El objetivo de este trabajo consistió en evaluar seis polimorfismos de nucleótido simple (SNPs) ubicados en los genes DGAT1, TG, LEP, GH, FABP4 y GnRHR como posibles marcadores genéticos apropiados para ser incluidos en un panel de trazabilidad para la identificación de la raza de origen en el contexto de la comercialización de carne bovina. Los resultados de la caracterización genética de cuatro de las principales poblaciones bovinas chinas y de las razas más importantes de nuestro país (cinco Bos taurus y dos B. indicus) sugieren que los marcadores estudiados pueden ser utilizados exitosamente como parte de un sistema de trazabilidad efectivo para identificar el origen de la carne bovina en el contexto de la importación de carne en el mercado chino y en particular en el comercio entre Argentina y China.
Palabras clave: Trazabilidad; Polimorfismo de nucleótido simple; Ganado chino; Ganado argentino; Comercialización.
INTRODUCTION
Nowadays, as a result of technology and globalization, food from all over the world can be found in the market, and meat is not an exception. Moreover, the world meat trade is expected to grow by 16% in 2020 compared with the average of 2008 - 2010, a demand growth that will stem mostly from large economies in Asia, Latin America and the oil exporting countries (OECD - FAO, 2011). In this scenario, China will probably keep its self-sufficiency policy, but due to its potential volumes both in terms of production and consumption, unforeseen events in China could result in a severe impact on the international markets (OECD - FAO, 2011). Besides, consumers from different countries or world regions have singular meat cut preferences that partly generate a trend of countries exchanging meat from the same breeds. Meat trade statistics show particular examples, like those of the US or Canada, which share a large part of their breeds and emerge in the first places as world meat exporters and importers at the same time (USDA, 2013). Consequently, there is a need to distinguish products of different origin due to aspects regarding authenticity (to avoid fraudulent products), quality (diverse productive systems or climates can result in different product merits) and safety (country/regional sanitary status).
During the last several years, consumers have increased their attention to food safety and quality due to the emergence of major diseases (e.g., Bovine spongiform encephalopathy -BSE-, avian influenza, food-borne diseases, etc.) or other health issues (e.g., the dioxin crisis that can generate serious threats to food safety. Furthermore, socio-economic reasons (e.g., changes in food habits towards healthier diets and increased consumption of organic food) have also contributed to the increase in consumer's interests in the origin, production methods, and industrialization processes of animal and plant products (Dalvit et al., 2007). As a consequence, issues associated with the traceability of food products have grown in importance, allowing consumers to consider food origin and processing in their purchasing decisions (Ajmone-Marsan et al., 2004).
It has been seen that consumers from different regions have different tastes and preferences in particular regarding meat cuts. Furthermore, quality attributes are different depending on the market, sometimes oriented to production processes (e.g. USDA natural meat), animal welfare (e.g. nutrition with GMO-free rations), and/or regulatory barriers (BSE or dioxin control). This results in a differentiation of meat products and creates a need of certification of guaranteed quality and safety of the beef production and the whole supply chain. In this sense, traceability has been a key issue to support the consumer's confidence. In particular, Argentine beef is well known all over the world for its superior quality, supported by the natural production system and the breeds raised (mostly British breeds). Consequently, a breed traceability system could complement the current system to guarantee that quality.
Several identification methods, including ear tags, ruminal bolus, retinal analysis, DNA markers, and tracking devices, have been evaluated to develop traceability systems. These methods allow product traceability at different levels: individual, geographical, breed and/or species (Negrini et al., 2008a; Negrini et al., 2008b; Li et al., 2009). Genetic traceability refers specifically to methods associated with the identification of animals and their products through DNA characterization of individuals, breeds or species. Methods for individual identification are mainly related to food safety, while discrimination between breeds and species is particularly useful to detect commercial frauds and to protect the value of local productions.
At present, microsatellites (STRs) are the markers most widely used for genetic identification and traceability studies (Peelman et al., 1998; Sancristobal-Gaudy et al., 2000; Arana et al., 2002; Vázquez et al., 2004; Herraeza et al., 2005; Dalvit et al., 2006; Orrú et al., 2006; Baldo et al., 2010) due to their high information degree provided by the large number of alleles that can be detected at each locus (Vignal et al., 2002; Dalvit et al., 2008). However, during the last few years, single nucleotide polymorphisms (SNPs) have become popular because they are based on the fundamental unit of genetic variation and are abundant across the genome. Moreover, SNPs have genetic stability, lower rates of genotyping error, and are amenable to automation and high-throughput genotyping technologies (Heaton et al., 2002; Heaton et al., 2005; Mariani et al., 2005; Werner et al., 2004, Karniol et al., 2009; Allen et al., 2010).
The characterization of a universal marker panel for breed traceability is complex because the most appropriate molecular markers for the specific breed groups that need to be assessed must be identified. The selection of markers will therefore depend on the gene frequency distribution, the genetic distance among breeds, and the presence of private alleles in target populations. To date, two major strategies have been used to characterize genetic traceability systems. One of them involves the use of STRs or SNPs, together with assignment tests (probabilistic strategy; Dalvit et al., 2007; Negrini et al., 2008a; Negrini et al., 2008b), whereas the other one is based on typing specific genes with private alleles (e.g., coat color genes), which eliminates the need for statistical inference (deterministic strategy; Ajmone-Marsan et al., 2004). Both strategies require, however, a preliminary characterization of the genetic structure of the populations under study.
In China, there are four Yellow cattle breeds that possess higher meat production capability than the other Chinese Yellow cattle breeds (Longworth et al., 2001). These four populations represent cattle from the north region and central agricultural region of China. Both Bos taurus and B. indicus as well as mixed breeds can be found within Chinese native cattle. Traditionally, the so-called Yellow cattle are classified into three groups: humpless, semi-humped, and humped types, coinciding with their distribution from the north to the south of China. In this sense, it has been demonstrated that cattle from the south and southwest have greater B. indicus influence (Jia et al., 2007) and that cattle from the northern region are related to European breeds (Sun et al., 2008). In the central agricultural region, cattle are mainly humped or semi-humped, as a result of northern Taurine and southern Zebuine crossbreeding (Sun et al., 2008). These breeds present complex patrilineages (Y-chromosome) and combined matrilineages (mtDNA) between B. taurus and B. indicus. During the last years, several commercial cattle breeds, including Holstein, Limousine and Simmental, have been introduced to China to improve dairy and beef production.
In Argentina, there are two main breeding areas: the temperate Pampa, where Holstein and British breeds, such as Angus and Hereford, are predominant, and the northeast subtropical region, where Zebuine and Creole cattle are the breeds most commonly raised. These four breeds were selected for the present study because they represent the main beef breeds raised in Argentina. Additionally, Wagyu, a valuable breed with specific meat quality raised in lower scale in Argentina, was included in the study (Rearte, 2007).
Considering the main features of Chinese and Argentine meat production, the aim of this study was to assess six SNPs located in candidate genes for meat quality as potential genetic markers to be included into a traceability panel for the identification of breed origin in the context of bovine beef trade. The candidate genes selected were: AcylCoA-diacylglycerol-acyltransferase 1 (DGAT1), Thyroglobulin (TG), Leptin (LEP), Growth hormone (GH), Fatty acid binding protein 4 (FABP4), and Gonadotropin releasing hormone receptor (GnRHR).
MATERIALS AND METHODS
Sample collection
Meat samples were collected in four Chinese commercial slaughterhouses from 80 individuals classified as Chinese Yellow cattle. One of the slaughterhouses was located in the north region (named herein Ch2) and three in the central agricultural region (named Ch1, Ch3 and Ch4 according to their northeast to southwest geographical location). In addition, blood samples were collected from 243 animals belonging to five populations of B. taurus breeds: Angus (AA), Hereford (HE), Holstein (HO), Wagyu (WA) and Argentine Creole (CR), and two B. indicus: Brahman (BR) and Nelore (NE). These populations represent the main breeds raised in the world and/or Argentina. In order to take a representative genetic profile of each breed, the samples were collected from several farms that comprise the different genetic lines present in each breed.
DNA extraction
Total DNA was extracted from blood samples using the Wizardâ Genomic DNA purification kit (Promega, Madison, WI, USA) following the manufacturer's instructions. DNA was also extracted from meat samples according to the methods previously reported by Wagner et al. (1994) and Giovambattista et al. (2001).
SNP genotyping
SNPs were genotyped by pyrosequencing methods, as described by Lirón et al. (2010) and Ripoli et al. (2011a). SNPs included K232A of DGAT1, GH6.1 of GH, R4C of LEP, I74V of FABP4, a T/C silent mutation in the fifth exon of the GnRHR gene, and the transition in the TG 5´ leader sequence (Grisart et al., 2002; Yao et al., 1996; Liefers et al., 2002; Hoashi et al., 2008; Barendse et al., 2001). Specific details of the SNPs analyzed are summarized in Table S1. The pyrosequencing assay comprised an initial PCR reaction of the target gene using one of the biotinylated primers. Amplified products were then purified by the streptavidin-coated Sepharose beads capture method to be used as pyrosequencing templates (Ronaghi, 2001). An internal sequencing primer, complementary to the biotinylated strand, was finally used to differentiate the allele variants of the target SNP. Pyrosequencing reactions were run on a PSQ96MA sequencer using the Pyro Gold Reagent Kit (Quiagen GmbH, Hilden, Germany) and analyzed using standard software for pyrosequencing genotyping (Biotage AB, Sweden).
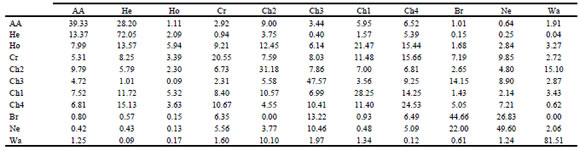
Table 1. Estimated gene frequencies for DGAT1, GH, LEP, FABP4, GnRHR and TG polymorphisms in Chinese Yellow cattle (named Ch1, Ch3 and Ch4), Angus (AA), Hereford (HE), Holstein (HO), Wagyu (WA) and Argentine Creole (CR), Brahman (BR) and Nelore (NE) breeds/populations. N = sample size.

Table S1. Details of the SNPs analyzed
.
Statistical analysis
Measures of genetic variability: The GENEPOP 4.0 software (Rousset, 2007) was used to calculate the allele frequencies for each SNP locus in all the populations studied. The observed (ho) and unbiased expected heterozygosity (he) for each locus and the average heterozygosity over all loci (He) were estimated according to Nei (Nei, 1978), using the ARLEQUIN 3.5 software for population genetic analyses (Schneider et al., 2000). Potential deviations from the Hardy-Weinberg equilibrium (HWE) were estimated by FIS statistics (Weir and Cockerman, 1984) and tested for each locus and population, as well as for all loci, using the exact test included in GENEPOP.
Genetic structure and population differentiation: Genetic structure and genetic differentiation among breeds were assessed through standard Wright's FST statistics, using the variance-based method of Weir and Cockerham (1984) and with the exact G test (Goudet et al., 1996) for population differentiation. These parameters were estimated using GENEPOP.
Levels of genetic differentiation between populations were described through population pairwise FST indices and represented graphically using the R-function: pairFstMatrix.r (Schneider et al., 2000). To assess the proportion of genetic variance explained by differences among and within breeds/populations, we initially performed an analysis of molecular variance (AMOVA) for all loci and for each individual locus considering all breeds as a single group. To estimate the proportion of genetic variance explained by individual breeds versus country of origin, we performed a hierarchical AMOVA, in which breeds/populations were grouped by their origin (i.e., European Taurine, Asiatic Taurine, and Zebuine). This analysis was also performed for each locus individually and for all loci. The AMOVA was carried out using ARLEQUIN. To condense the genetic variability revealed by the six SNPs, allele frequencies were used to perform a Principal Components Analysis (PCA) according to Cavalli-Sforza et al. (1994) and implemented using the PAST software (Hammer et al., 2001).
Nei´s standard genetic distance (DS; Nei, 1972) was calculated from allele frequencies to perform a cluster analysis using the unweighted pair group method with arithmetic mean (UPGMA, Sneath and Sokal, 1973) algorithm. Confidence for the groupings was estimated through bootstrap resampling of the data using 1000 replications. Genetic distances and trees were computed using the POPULATIONS 1.2.28 software (Langella, 1999). The trees were then visualized using TREEVIEW (Page, 1996).
Assignment test: Assignment tests were performed using simulations of 10000 multilocus genotype data generated from gene frequencies from each of the eleven breeds, assuming Hardy-Weinberg and linkage equilibrium (Paetkau et al., 1995; Liron et al., 2007). This assignment test involves calculating the expected frequency of each individual's genotype in each of the breeds/populations studied and, subsequently, estimating assignment probabilities of each individual to the population where its expected genotype frequency was the highest. Finally, we calculated the percentage of samples correctly assigned to each breed.
RESULTS
Estimates of genetic variability within breeds
Allele frequencies for all breeds are presented in Table 1. The DGAT1-K and FABP4-C variants had higher allele frequencies in Zebuine and Ch3 breeds. In contrast, the DGAT1-A allele was the most abundant variant in Taurine breeds. The FABP4-T and -C alleles exhibited similar incidence in Taurine breeds except HE, which showed a high frequency for FABP4-T. For GnRHR, both allele variants had similar frequencies in most of the Taurine breeds, while GnRHR-A was the most common allele in CR, WA, Ch2 and Zebuine breeds. TG-C and GH-C alleles showed relatively high frequencies in all the breeds studied except WA. The LEP-A allele was the most abundant in the British breeds, while the LEP-G allele showed higher frequencies in all the other breeds.
From a total of 66 locus-population possible combinations, we were able to perform 64 HWE tests. Two locus-population combinations were excluded because of limited polymorphism; one allele variant was fixed for locus GH in the BR population and a second allele was almost fixed with TG at very high frequency (Minimum Allele Frequency, MAF < 0.013) in the HE population. HWE tests revealed that only five locus-population combinations were statistically significant (P = 0.05) (Table 2). These deviations corresponded to single-locus differences in HE, CR, WA, Ch3 and Ch1. Non-significant deviations from HWE were observed for the other six breeds analyzed.
Table 2. Observed (ho) and expected (he) heterozygosities, average heterozygosity (He), significant FIS index and FST for six SNPs in Chinese Yellow cattle (named Ch1, Ch3 and Ch4), Angus (AA), Hereford (HE), Holstein (HO), Wagyu (WA) and Argentine Creole (CR), Brahman (BR) and Nelore (NE) breeds/populations.

nd = not determined.
Table S2. Percentage of individual assignment. Chinese Yellow cattle (named Ch1, Ch3 and Ch4), Angus (AA), Hereford (HE), Holstein (HO), Wagyu (WA) and Argentine Creole (CR), Brahman (BR) and Nelore (NE).
Estimates of observed (ho) and unbiased expected (he) heterozygosities for each locus and breed studied are shown in Table 2. The expected heterozygosity ranged from 0.026 for TG in the HE breed to 0.508 for GnRHR in the Ch1 population and for FABP4 in the CR, WA, HO, Ch1 and Ch4 breeds/populations. The average heterozygosity (He), also estimated for each population, ranged from 0.261 in NE to 0.455 in Ch2 cattle.
Genetic structure and levels of population differentiation
The FST index and the exact G test for population differentiation were used to analyze the degree of genetic differentiation among the cattle breeds studied. FST showed significant differences across all cattle populations (FST = 0.1530), ranging from 0.056 to 0.260 for each individual locus (Table 2 and Figure S1). The exact G test for population differentiation indicated that differences in allele frequency distributions among populations were highly significantly (exact P value for all loci = 0.0001).

Figure 1. The unweighted pair group method with arithmetic mean (UPGMA) tree was constructed from a matrix of Nei´s standard (Ds) genetic distances using the allele frequencies of the DGAT1, GH, LEP, FABP4, GnRHR and TG polymorphisms in Chinese Yellow cattle (named Ch1, Ch3 and Ch4), Angus (AA), Hereford (HE), Holstein (HO), Wagyu (WA) and Argentine Creole (CR), Brahman (BR) and Nelore (NE).
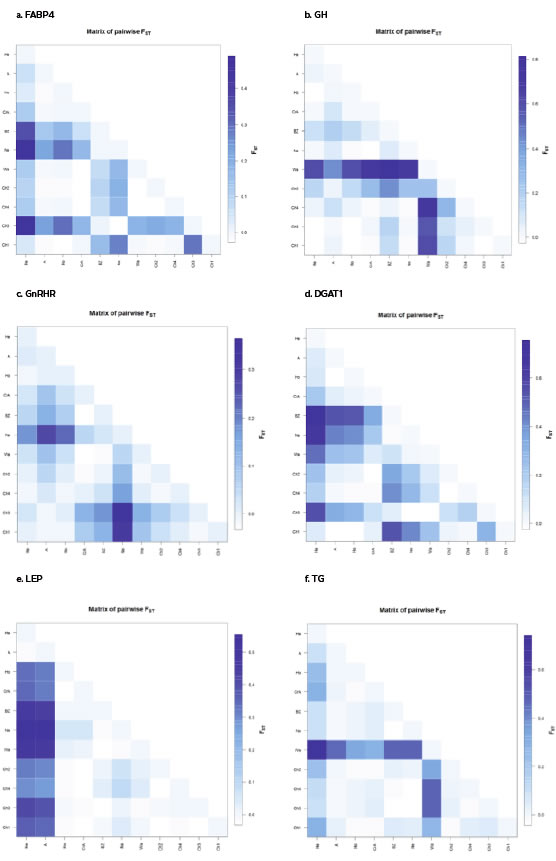
Supplementary figure S1. Graphic representation of calculated FST between population pairs using an R-function: pairFstMatrix.r.
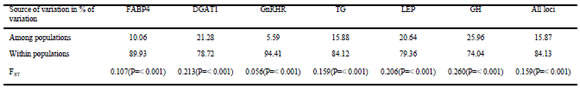
The overall AMOVA, which considered all breeds as a single group, revealed that 15.87% of the genetic variance observed was explained by differences among populations, whereas the other 84.13% was explained by differences among individuals within populations. The hierarchical AMOVA allowed partitioning the genetic variability in different breed groups based on their historical origin. When breeds were grouped in three clusters according to their origin, the AMOVA showed that differences among groups accounted for 8.75% of the total genetic variance, while differences among populations within groups accounted for 9.62%. As with the overall AMOVA, most of the variation (81.63%) was explained by differences among individuals.
The AMOVAs were also performed for each individual locus, considering all breeds as a single group. This analysis revealed a wide range of variation in the proportion of variance explained by differences among populations (5.59 - 25.96%), with the majority of the variation explained by differences among individuals within populations (74.04 - 94.41%, Table 3). The highest percentage of variation among populations was detected for the GH gene, with 25.96% of the variation being explained by allele frequencies at this locus. GnRHR was the less informative locus regarding population structuring, with only 5.59% of the variation explained by differences among populations.
Table 3. Percentage of variation for each individual polymorphism (DGAT1, GH, LEP, FABP4, GnRHR and TG) and all loci obtained by AMOVA considering all breeds/populations (Chinese Yellow cattle, Angus, Hereford, Holstein, Wagyu, Argentine Creole, Brahman and Nelore) as a single group.
When populations were grouped according to their European, Asiatic or Zebuine origin, the hierarchical AMOVA also revealed considerable levels of variation within populations (73.31 to 92.56%). However, the genetic variance among groups accounted for up to 19.56% of the genetic diversity, depending on the genetic marker considered (0.72 to 19.56%), whereas the variance among populations within each group explained 4.53 to 24.18% of the total genetic variance.
Allele frequencies were used to generate the DS for each pair including the 11 cattle populations studied. A distance matrix based on the UPGMA algorithm was used to perform a cluster analysis and assess relationships between cattle breeds. The UPGMA cluster analysis using the DS was consistent with the historical and geographical origin of population/breeds. The tree revealed the main divergences separating the Asiatic Taurine, European Taurine and Zebuine clusters (Figure 1).
Principal Components Analysis (PCA)
Results from the PCA are reported in Figure 2, which illustrates the first and second Principal components (PCs) for the six SNPs frequency distributions of the populations/breeds analyzed. The first two components cumulatively accounted for 84.54% of the variability in the data. The first PC accounted for 51.24% of the total variance and showed a differentiation pattern between Zebuine breeds (NE and BR) and British breeds (AA and HE). These breeds were mainly differentiated by the allele frequencies at the DGAT1, LEP and FABP4 loci and, to less extent, by the GnRHR gene frequencies. The second PC explained 33.30% of the variation and clearly distinguished WA from all the other populations. The second PC was explained mainly by differences in allele frequencies at the GH and TG loci. Although the third PC accounted for 8.53% of the variance, it provided no information regarding the potential origin of the breeds. PCA results were also consistent with the overall results of the cluster analyses generated using the UPGMA algorithm.

Figure 2. Principal Components Analysis (PCA) of the allele frequencies of the DGAT1, GH, LEP, FABP4, GnRHR and TG polymorphisms in Chinese Yellow cattle (named Ch1, Ch3 and Ch4), Angus (AA), Hereford (HE), Holstein (HO), Wagyu (WA) and Argentine Creole (CR), Brahman (BR) and Nelore (NE) breeds/populations.
Assignment test
An assignment test was performed using the simulated multilocus genotype data. The results for correct allocation percentage were variable among breeds, ranging from 5.94% in HO to 81.51% in WA. However, for all the breeds except HO, the highest percentage of individual assignment within each breed matched to its own population (Table S2). In general, the highest percentage of wrong allocation corresponded to more closely related breeds. For example, AA was confused with HE and NE was confused with BR. These results are in agreement with the grouping pattern obtained with PCA. An assignment test considering groups of breeds by their origin was calculated and is shown in Table 4.
Table 4. Percentage of correct assignment grouped by breed origin: British (Angus and Hereford), HO (Holstein), CR (Argentine Creole), Chinese Yellow cattle, Zebuine (Brahman and Nelore), and WA (Wagyu).

DISCUSSION AND CONCLUSIONS
A genetic traceability system is needed to ensure certification of breed origin and beef quality. The first step in the implementation of a genetic traceability system to certify the breed origin of beef products involves the identification of an appropriate set of molecular markers with significant levels of variation that can be explained by group origin, and the genetic characterization of the breed/populations involved in the assessment. In the context of China's beef trading, we assessed the four main beef cattle populations raised in this country and four of the main breeds raised worldwide, to evaluate the feasibility of certifying the origin of breeds or breed products and detecting potential commercial frauds through a genetic traceability system. Furthermore, to certify high quality Argentine beef, mainly from British cattle breeds (such as Argentine AnGus Beef and Argentine Hereford Beef trademarks), HO and Zebuine breeds were included as possible sources of fraud. In addition, beef from WA, a more exotic breed with high marbling grade, was also included because it is produced in Argentina for exportation to premium markets.
The effectiveness of a traceability system will depend greatly on the levels of polymorphisms, allele frequency distributions, and genetic differentiation detected among the breeds under consideration, as well as on the potential presence of private alleles (i.e., breed-specific alleles). In bovines, most differences among breeds are due to differences in allele frequency distributions rather than to the presence of private alleles. As a consequence, most systems require probabilistic methods of traceability, associated with population assignment probabilities, rather than deterministic methods based on population-specific genetic variants. Within the group of SNPs selected in this study, mostly candidate genes for fat content, there were no private alleles, but the gene frequencies showed significant levels of variation across the breeds studied, accounting for 15.87% of the genetic variance.
The results obtained show that the SNPs analyzed separate WA from the other breeds, and divide the remaining populations into three groups: Taurine, mixed and Zebuine breeds. GH and TG loci evidenced that the gene frequency profile in WA was notably different from that in the other breeds, as previously observed (Ripoli et al., 2011b), which could be explained by the high level of selection done on WA to increase marbling in this breed (Zembayashi and Lunt, 1995). In the DGAT1 locus, the allele frequencies evidenced a geographical cline given by the high frequencies for the A allele in European (fixed in HE), Ch1 and Ch4 populations followed by intermediate values in CR, Ch2, WA and lastly by Ch3, NE and BR breeds. These results are also concordant with the degree of Zebu admixture expected within each breed/population analyzed and are in agreement with previous reports (Winter et al., 2002; Ripoli et al., 2006). In the case of the FABP4 gene, a similar gradient was also observed. The FABP4-T variant showed intermediate frequencies in European and Asiatic Taurine breeds, and lower frequencies in Ch3 and Zebuine breeds. Since Cho et al. (2008) reported that the I74V FABP4 polymorphism was associated positively with back fat thickness in Korean cattle, our results could be related to those of Cho et al. (2008) because Zebuine breeds are leaner than Taurine ones. The similar frequencies in Ch3, NE and BR may result from the strong influence of Zebu breeds on some populations of the central agricultural region of China, especially on the Ch3 population. Regarding the LEP gene, the LEP-G variant exhibited a high frequency in almost all the breeds analyzed, but a low frequency in the British ones. In the GnRHR locus, although Zebuine breeds showed higher allele frequencies for the GnRHR-A variant, we could not detect a clear pattern of frequency distribution. Summarizing, GH and TG gene frequencies seem to be useful to distinguish WA cattle from the other breeds. DGAT1 and FABP4 differentiated between Taurine, mixed and Zebuine breeds, as they present a gradient of frequencies, while LEP differentiated the British breeds from the rest. All these markers showed FST values higher than 0.1, which is enough to allow an efficient differentiation of breeds or breed groups. These FST values were compared with those previously reported using neutral STR markers in the same populations (Liron et al., 2007; Rogberg et al., 2012). The SNP FST values ranged from 0.107 to 0.26, while the STR FST values ranged between 0.069 and 0.151. These results evidenced that the SNPs analyzed show greater population structuring than STRs. This fact might be the consequence of selective forces, and supports the hypothesis of the present work that the SNP markers associated with meat quality can be successfully used as a part of an effective traceability system for the identification of cattle breed origin. Regarding GnRHR, it seems to be less useful for breed traceability in the Chinese-Argentine beef trade context because it showed a low degree of genetic differentiation among the breeds studied.
Additionally, the tree constructed with UPGMA, using DS, reflected the same results. The WA breed was located at one end of the tree, and the Zebuine group (NE and BR) and Ch3 population were located at the other end. The British breeds clustered together and the rest of the breeds/populations were placed in an intermediate position between the WA and Zebuine breeds. The tree exhibits a topology consistent with the historical and geographical origin, combined with a selection towards body and/or meat fat content in the population/breeds analyzed. Moreover, the results observed in terms of the PCA also matched with the topology of the tree. In this sense, differences in frequency distribution provide the basis to characterize individual breeds through cluster analysis and PCA.
Finally, an assignment test was performed to validate the effectiveness of the SNPs analyzed to differentiate beef of the main breeds raised in the world and/or Argentina (AA, HE, NE and BR) from premium beef (WA) of those breeds typically raised in China for beef purposes (native breeds and HO). Marker sets previously proposed for breed assignment included more than 11-30 STRs or 25-90 SNPs (Dalvit et al., 2007, 2008; Negrini et al., 2008a,b; Karniol et al., 2009; Baldo et al., 2010). However, although only six SNPs were analyzed in the present work, they evidenced reasonable values of correct allocation. These values are particularly higher (> 70%) for the high quality beef breeds (AA, HE, WA) and lower for the low quality ones (HO, NE and BR). The observed results are in agreement with those obtained with PCA and with the topologies of the tree. The breeds that exhibited extreme values for the first and second PC presented higher percentage of correct allocations. By contrast, breeds with values closer to 0 for the first and second PC showed higher values of wrong allocations. In the case of Argentine high quality beef (AA)
REFERENCES
1. Ajmone-Marsan P., Milanesi E., Negrini R. (2004) Breed traceability using molecular methods. In Proceedings of the 7th World Conference of the Brown Swiss Cattle Breeders (pp. 101-104). [ Links ]
2. Allen A.R., Taylor M., McKeown B., Curry A.I., Lavery J.F., Mitchell A., Hartshorne D., Fries R., Skuce R.A. (2010) Compilation of a panel of informative single nucleotide polymorphisms for bovine identification in the Northern Irish cattle population. BMC Genet. 11: 5. [ Links ]
3. Arana A., Soret B., Lasa I., Alfonso L. (2002) Meat traceability using DNA markers: Application to the beef industry. Meat. Sci. 61:367-373. [ Links ]
4. Baldo A., Rogberg-Muñoz A., Prando A., Mello Cesar A.S., Lirón J.P., Sorarrain N., Ramelli P., Posik D.M., Pofcher E., Ripoli M.V., Beretta E., Peral-García P.,Vaca R., Mariani P., Giovambattista G. (2010) Effect of consanguinity on Argentinean Angus beef DNA traceability. Meat. Sci. 85(4): 671-675. [ Links ]
5. Barendse W., Bunch R., Thomas M., Armitage S., Baud S., Donaldson N. (2001) The TG5 DNA marker test for marbling capacity in Australian feedlot cattle. Livestock Library com.au. http://www.beef.crc.org.au/Publications/MarblingSym/Day1/Tg5DNA. (Accessed 9 March 2003). [ Links ]
6. Cavalli-Sforza L.L., Menozzi P., Piazza A. (1994) The history and geography of human genes. Princeton University Press, Princeton, New Jersey [ Links ]
7. Cho S., Park T.S., Yoon D.H., Cheong H.S., Namgoong S., Park B.L., Lee H.W., Han C.S., Kim E.M., Cheong I.C., Kim H., Shin H.D. (2008) Identification of genetic polymorphisms in FABP3 and FABP4 and putative association with backfat thickness in Korean native cattle. BMB Reports 31, 41(1): 29-34. [ Links ]
8. Dalvit C., De Marchi M., Cassandro M. (2007) Genetic traceability of livestock products: A review. Meat Sci. 77: 437-449. [ Links ]
9. Dalvit C., De Marchi M., Targhetta C., Gervaso M., Cassandro M. (2008) Genetic traceability of meat using microsatellite markers. Food Res. Int. 41: 301-307. [ Links ]
10. Dalvit C., Targhetta C., Gervaso M., De Marchi M., Mantovani R., Cassandro M. (2006) Application of a panel of microsatellite markers for the genetic traceability of bovine origin products. In Proceedings of 57th Annual Meeting of the European Association for Animal Production (pp. 26). [ Links ]
11. Giovambattista G., Ripoli M.V., Lirón J.P., Villegas-Castagnaso E.E., Peral-García P., Lojo M.M. (2001) DNA typing in a cattle stealing case. J. Forensic. Sci. 46: 1484-1486. [ Links ]
12. Goudet J., Raymond M., Demeeüs T., Rousset F. (1996) Testing genetic differentiation in diploid populations. Genetics 144: 1933-1940. [ Links ]
13. Grisart B., Coppieters W., Farnir F. (2002) Positional candidate cloning of a QTL in dairy cattle: identification of a missense mutation in the bovine DGAT1 gene with major effect on milk yield and composition. Genome Res. 12: 222-231. [ Links ]
14. Hammer Ø., Harper D.A.T., Ryan P.D. (2001) PAST: paleontological statistics software package for education and data analysis. Palaeontological Electronica 4. http://palaeo-electronica.org/2001_1/past/issue1_01.htm. [ Links ]
15. Heaton M.P., Harhay G.P., Bennett G.L., Stone R.T., Grosse W.M., Casas E., Keele J.W., Smith T.P.L., Chitko-McKown C.G., Laegreid W.W. (2002) Selection and use of SNP markers for animal identification and paternity analysis in U.S. beef cattle. Mamm. Genome. 13(5): 272-281. [ Links ]
16. Heaton M.P., Keen J.E., Clawson M.L., Harhay G.P., Bauer N., Schultz C., Green B.T., Durso L., Chitko-McKown C.G., Laegreid W.W. (2005) Use of bovine single nucleotide polymorphism markers to verify sample tracking in beef processing. J. Am. Vet. Med. Assoc. 226(8): 1311-1314. [ Links ]
17. Herraeza D.L., Schafer H., Mosner J., Fries H.R., Wink M. (2005) Comparison of microsatellite and single nucleotide polymorphism markers for the genetic analysis of a Galloway cattle population. Z. Naturforsch. C. 60(7-8): 637-643. [ Links ]
18. Hoashi S., Hinenoya T., Tanaka A., Ohsaki H., Sasazaki S., Taniguchi M., Oyama K., Mukai F., Mannen H. (2008) Association between fatty acid compositions and genotypes of FABP4 and LXR-alpha in Japanese Black cattle. BMC Genet. 9: 84. [ Links ]
19. Jia S., Chen H., Zhang G., Wang Z., Lei C., Yao R., Han X. (2007) Genetic variation of mitochondrial D-loop region and evolution analysis in some Chinese cattle breeds. J. Genet. Genomics 34(6): 510-518. [ Links ]
20. Karniol B., Shirak A., Baruch E., Singrün C., Tal A., Cahana A., Kam M., Skalski Y., Brem G., Weller J.I., Ron M., Seroussi E. (2009) Development of a 25-plex SNP assay for traceability in cattle. Anim. Genet. 40(3): 353-356. [ Links ]
21. Langella O. (1999) POPULATIONS version 1.2.28. Population genetic software. http://www.pge.cnrs-gif.fr. [ Links ]
22. Li Y., Wei Y.M., Pan J.R., Guo B.L. (2009) Determination of geographical origin of beef based on FTIR spectroscopy analysis. Guang Pu Xue Yu Guang Pu Fen Xi 29(3): 647-651. [ Links ]
23. Liefers S.C., te Pas M.F., Veerkamp R.F., van der Lende T. (2002) Associations between leptin gene polymorphisms and production, live weight, energy balance, feed intake, and fertility in Holstein heifers. J. Dairy Sci. 85: 1633-1638. [ Links ]
24. Lirón J.P., Prando A., Ripoli M.V., Rogberg-Muñoz A., Posik D.M., Baldo A., Peral-García P., Giovambattista G. (2011) Characterization and validation of bovine Gonadotropin releasing hormone receptor (GNRHR) polymorphisms. Res. Vet. Sci. 91(3):391-6. [ Links ]
25. Lirón J.P., Ripoli M.V., Peral-García P., Giovambattista G. (2007) Implication of population structure in the resolution of cattle stealing cases. J. Forensic Sci. 52(5): 1077-1081. [ Links ]
26. Longworth J.W., Brown C.G., Waldron S.A. (2001) Beef in China: agribusiness, opportunities and challenges. University of Queensland Press, St. Lucia, Australia. [ Links ]
27. Mariani P., Panzitta F., Nardelli Costa J., Lazzari B., Crepaldi P., Marilli M., Fornarelli F., Fusi M., Milanesi E., Negrini R., Silveri R., Filippini F., Ajmone Marsan P. (2005) Metodi molecolari per la tracciabilita dei prodotti di origine animale. In Proceedings of the 4th World Italian Beef Cattle Congress (pp. 297-302). [ Links ]
28. Negrini R., Nicoloso L., Crepaldi P., Milanesi E., Colli L., Chegdani F., Pariset L., Dunner S., Leveziel H., Williams J.L., Ajmone Marsan P. (2008a) Assessing SNP markers for assigning individuals to cattle populations. Anim. Genet. 40: 18-26. [ Links ]
29. Negrini R., Nicoloso L., Crepaldi P., Milanesi E., Marino R., Perini D., Pariset L., Dunner S., Leveziel H., Williams J.L., Ajmone Marsan P. (2008b) Traceability of four European Protected Geographic Indication (PGI) beef products using Single Nucleotide Polymorphisms (SNP) and Bayesian statistics. Meat. Sci. 80: 1212-1217. [ Links ]
30. Nei M. (1972) Genetic distance between populations. Am. Nat. 106: 283-292. [ Links ]
31. Nei M. (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89: 583-590. [ Links ]
32. Orrù L., Napolitano F., Catillo G., Moioli B. (2006) Meat molecular traceability: How to choose the best set of microsatellites? Meat Sci. 72: 312-317. [ Links ]
33. Paetkau D., Calvert W., Stirling I., Strobeck C. (1995) Microsatellite analysis of population structure in Canadian polar bears. Mol. Ecol. 4: 347-354. [ Links ]
34. OECD-FAO Agricultural Outlook 2012-2021 (2012) http://www.oecd.org/site/oecd-faoagriculturaloutlook (Accessed 11th October 2012). [ Links ]
35. Page R.D.M. (1996) TREEVIEW: an application to display phylogenetic trees on personal computers. Comput. App. Biosci. J. 12:357-358. [ Links ]
36. Peelman L.J., Mortiaux F., Van Zeveren A., Dansercoer A., Mommens G., Coopman F., Bouquet Y, Burny A, Renaville R, Portetelle D. (1998) Evaluation of the genetic variability of 23 bovine microsatellite markers in four Belgian cattle breeds. Anim. Genet. 29 (3): 161-167. [ Links ]
37. Rearte D. (2007) La producción de carne en Argentina. URL: http://www.inta.gov.ar/balcarce/Carnes/ProdCarneArg_esp.pdf (07-09-10). [ Links ]
38. Ripoli M.V., Corva P., Giovambattista G. (2006) Analysis of a polymorphism in the DGAT1 gene in 14 cattle breeds through PCR-SSCP methods. Res. Vet. Sci. 80: 287-290. [ Links ]
39. Ripoli M.V., Rogberg-Muñoz A., Liron J.P., Giovambattista G. (2011a) Development of typing methods based on pyrosequencing technology for the analysis of six bovine genes related to marbling. Journal of Basic and Applied Genetics (in press). [ Links ]
40. Ripoli M.V., Rogberg-Muñoz A., Lirón J.P., Francisco E., Villegas-Castagnasso E.E., Peral-Garcia P., Giovambattista G. (2011b) History and selection imprinting on genetic relationships among bovine breeds analyzed trough five genes related with marbling. Res. Vet. Sci. 90(2): 245-252. [ Links ]
41. Rogberg-Muñoz A., Wei S., Ripoli M.V., Guo B., Goszczynski D.E., Carino M., Castillo N.S., Melucci L., Villareal E., Liron J.P., Crespi J.A., Wei Y., Giovambattista G. Evaluation of STR set for bovine traceability in the context of Chinese Beef Imports and Argentine-Chinese beef trade. Comunicaciones Libres XXXIII Conference of the International Society of Animal Genetics, 15-20 Julio 2012, Cairns, Australia; p. 111. [ Links ]
42. Ronaghi M. (2001) Pyrosequencing sheds light on DNA sequencing. Genome Res. 11: 3-11. [ Links ]
43. Rousset F. (2007) Inferences from spatial population genetics. In: Handbook of statistical genetics. Eds. D. J. Balding, M. Bishop & C. Cannings, pp. 945-979. Wiley, Chichester, U.K., 3rd edn. [ Links ]
44. Sancristobal-Gaudy M., Renand G., Amigues Y., Boscher M.Y., Leveziel H., Bibé B. (2000) Traçabilté individuelle des viandes bovine à l'aide de marqueurs génétiques. INRA Prod. Anim. 13(4): 269-276. [ Links ]
45. Schneider S., Roessli D., Excoffier L. (2000) Arlequin Version 2.000: A software for population genetics data analysis. Genetics and Biometry Laboratory, University of Geneva, Switzerland. [ Links ]
46. Sevane N., Crespo I., Cañón J., Dunner S. (2011) A Primer-Extension Assay for simultaneous use in cattle Genotype Assisted Selection, parentage and traceability analysis. Livestock Science 137: 141-150. [ Links ]
47. Sneath P.H.A., Sokal R.R. (1973) Numerical taxonomy. San Francisco, CA: W. H. Freeman. [ Links ]
48. Sun W., Chen H., Lei C., Lei X., Zhang Y. (2008) Genetic variation in eight Chinese cattle breeds based on the analysis of microsatellite markers. Genet. Sel. Evol. 40(6): 681-692. [ Links ]
49. USDA (United States Department of Agriculture), Livestock and Poultry: World Markets and Trade (2013) http://www.fas.usda.gov (Accessed 23th May 2013). [ Links ]
50. Vázquez J.F., Pérez T., Ureña F., Gudín E., Albornoz J., Domínguez A. (2004) Practical application of DNA fingerprinting to trace beef. J. Food Prot. 67(5): 972-979. [ Links ]
51. Vignal A., Milan D., San Cristobal M., Eggen A. (2002) A review on SNP and other types of molecular markers and their use in animal genetics. Genet. Sel. Evol. 34: 275-305. [ Links ]
52. Wagner V., Schild T., Geldermann H. (1994) DNA variants within the 5-flanking regions of milk protein genes. II. The b-lactoglobulin-encoding gene. Theor. Appl. Genet. 89: 121. [ Links ]
53. Weir B.S., Cockerham C.C. (1984) Estimating F-statistics for the analysis of populations structure. Evolution 38: 1358-1370. [ Links ]
54. Werner F.A.O., Durstewitz G., Habermann F.A., Thaller G., Kramer W., Kollers S., Buitkamp J., Georges M., Brem G., Mosner J., Fries R. (2004) Detection and characterization of SNP useful for identity control and parentage testing in major European dairy breeds. Anim. Genet. 35: 44-49. [ Links ]
55. Winter A., Krämer W., Werner F.A., Kollers S., Kata S., Durstewitz G., Buitkamp J. Womack J.E., Thaller G., Fries R. (2002) Association of a lysine-232/alanine polymorphism in a bovine gene encoding acyl-CoA:diacylglycerol acyltransferase (DGAT1) with variation at a quantitative trait locus for milk fat content. Proceedings of the National Academy of Sciences of the United States of America 99(14): 9300-9305. [ Links ]
56. Yao J., Aggerey S.E., Zadworny D., Hayes J.E., Kuhnlein U. (1996) Sequence variations in the bovine growth hormone gene characterized by single strand conformation polymorphism (SSCP) analysis and their association with milk production traits in Holstein. Genetics 144: 1809-1816. [ Links ]
57. Zembayashi M., Lunt D.K. (1995) Distribution of intramuscular lipid throughout M. longissimus thoracis et lumborum in Japanese Black, Japanese Shorthorn, Holstein and Japanese Black crossbreds. Meat. Sci. 40(2): 211-216. [ Links ]

