










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkBAG. Journal of basic and applied genetics
versión On-line ISSN 1852-6233
BAG, J. basic appl. genet. vol.25 no.2 Ciudad Autónoma de Buenos Aires dic. 2014
ARTÍCULOS ORIGINALES
Componentes principales en mapeo asociativo
Principal components in associative mapping
Peña Malavera A.1, Gutierrez L.2, Balzarini M.1
1 Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET). Estadística y Biometría, Facultad de Ciencias Agropecuarias, Universidad Nacional de Córdoba, Av. Valparaíso s/n, Ciudad Universitaria, CP: 5000 (509) Córdoba, Argentina.
2 Facultad de Agronomía, Universidad de la República mbalzari@agro.unc.edu.ar
Fecha de recepción: 09/06/2014
Fecha de aceptación de versión final: 25/06/2014
RESUMEN
El mapeo asociativo (o mapeo por desequilibrio de ligamiento) permite encontrar lugares específicos del genoma relacionados con la variación de un carácter fenotípico. Es una práctica difundida en el mejoramiento de especies vegetales ya que no necesita la utilización de poblaciones provenientes de cruzamientos controlados. Sin embargo, se ha detectado que en poblaciones estructuradas genéticamente, el número de falsos positivos en la asociación marcador-carácter puede aumentar significativamente. El análisis de componentes principales constituye una herramienta para identificar la estructura y expresar la misma en un número reducido de componentes principales (CPs). Entonces, estos CPs se pueden incorporar como covariables en el modelo de asociación. Diferentes estrategias de modelado se pueden utilizar para tener en cuenta la estructura genética en el mapeo de asociación. El objetivo de este trabajo fue estimar las tasas de falsos positivos derivados de la aplicación de tres modelos estadísticos diferentes de mapeo bajo poblaciones estructuradas. Los modelos comparados fueron M1: sin corrección por estructura, M2: con las CPs como covariables de efectos fijos y M3: incluyendo las mismas CPs como covariables de efectos aleatorios en el marco de un modelo lineal mixto. La comparación se realizó usando datos reales y simulados de una especie autógama. Los resultados sugieren que la corrección con las CPs como covariables aleatorias disminuye la tasa de falsos positivos en la inferencia de asociaciones marcador-carácter.
Palabras clave: Modelos lineales mixtos; Análisis de componentes principales; Estructura genética.
ABSTRACT
Association mapping (or linkage disequilibrium mapping) is used to find specific parts of the genome associated with phenotypic trait variation. It is a widely used in plant breeding because it allows the use of populations that do not come from specific experimental designs. If the population of individuals used in association mapping is genetically structured, the number of false positives, in the marker-trait association, increases. Several strategies can be used to model associations taken into account the underlying genetic structure. The principal components analysis can be used to identify the structure and express it in a reduced number of principal components (PCs). Then, these PCs can be incorporated as covariates in the association model. Different models strategies can be used to account for genetic structure in association mapping. The aim of this paper is to estimate expected false positive rates in association mapping performed by three different statistical models, under genetically structured populations. Compared models were M1: without correction for structure, M2: including PCs, as covariates of fixed effects, and M3: including PCs as random effects within a linear mixed model. Model comparison was performed using both, real and simulated data, for self-pollinated specie. The results suggested that the use of PCs as random covariates decreases the false positive rate in the inference of marker-trait associations.
Key words: Mixed linear models; Principal components analysis; Genetic structure.
INTRODUCCIÓN
El mapeo asociativo, también conocido como mapeo por desequilibrio de ligamiento (linkage disequilibrium, LD), permite identificar lugares específicos del genoma relacionados con la variación de un carácter fenotípico de interés. La técnica nace en el contexto del estudio de enfermedades en humanos por la imposibilidad de desarrollar poblaciones provenientes de cruzamientos controlados (Corder, 1994). En los últimos años se ha incrementado su uso para la identificación de genes responsables de características complejas de interés agronómico y actualmente esta práctica se encuentra difundida y adoptada en el mejoramiento de especies vegetales para el análisis de los loci involucrados con caracteres cuantitativos (QTL) (Remington et al., 2001; Kraakman et al., 2006; Aranzana et al., 2005; Breseghello y Sorrells, 2006; D'hoop et al., 2008; Stich et al., 2008; Thornsberry et al., 2001; Zhu et al., 2008).
El LD puede definirse como el grado de asociación no aleatoria entre alelos de distintos loci en poblaciones de individuos no relacionados (Yu y Buckler, 2006); se relaciona con la proporción de gametos que no segregan al azar y provee información sobre la historia de la población así como sobre el sistema de selección implementado. A nivel genómico, refleja el impacto de fuerzas evolutivas (selección natural, mutación y migración) que causan cambios en las frecuencias génicas (Lynch y Walsh, 1998; Falconer y Mackay, 1996).
Cuando la población de individuos empleada en el mapeo por LD está estructurada genéticamente, aumenta la cantidad de falsos positivos en la detección de las asociaciones de interés (Malosetti et al., 2007). Esto ocurre porque en una población con sub-poblaciones, cualquier carácter presente con mayor frecuencia en una de ellas mostrará asociación positiva con alelos que son más comunes en esta sub-población (Zhang et al., 2010). Consecuentemente, es posible que se detecten marcadores asociados con la composición de la población más que con la característica de interés (Yu et al., 2006). Esta situación aumenta la tasa de falsos positivos. Por ello, se han propuesto distintas estrategias de mapeo asociativo tendientes a controlar el aumento en la detección de asociaciones espurias. Una estrategia es incluir en el modelado, covariables que representen la estructura genética.
Una forma de estudiar la estructura subyacente es estimar la probabilidad de pertenencia de cada individuo a las sub-poblaciones que componen la meta-población (Pritchard et al., 2000). La información de la clasificación resultante se puede incorporar al modelo de mapeo de distintas formas (Yu et al., 2006; Gutiérrez et al., 2011). Price et al. (2006) proponen contemplar la estructura genética a través de una matriz compuesta por variables sintéticas resultantes de un análisis de componentes principales (ACP) (Hottelling, 1936) realizado sobre la matriz de datos de marcadores genéticos. El ACP es una técnica de reducción de dimensión, ampliamente usada en diversas áreas de la biología e implementada en la mayoría de los programas estadísticos. Con el ACP es posible obtener un conjunto de nuevas variables, generadas como combinación lineal de las variables originales. Estas variables sintéticas o componentes principales (CPs) tienen la característica de ser no correlacionadas y óptimas para señalar variabilidad o diferencias entre los casos en estudio. Las CPs, conformadas a través de la combinación de marcadores moleculares de los genotipos de la población de mapeo permiten señalar diferencias entre genotipos causadas por la existencia de estructuración genética. Así las CPs significativas, según la prueba de Tracy y Widom (1994), han sido usadas como covariables en el modelo de mapeo asociativo (regresión lineal múltiple) (Wang et al., 2012; Cappa et al., 2013; Muñoz-Amatriaín et al., 2014). Tales covariables pueden contemplarse en el modelo de mapeo como efectos fijos o aleatorios, para este último caso la estimación se realizó en el contexto de un modelo lineal mixto (MLM) (Searle, 2008). Malosetti et al. (2007) recomiendan incluir la estructura genética subyacente en las poblaciones de mapeo como efecto aleatorio, independientemente del procedimiento utilizado para detectar dicha estructura y del nivel de LD subyacente. Sin embargo, no se conoce si tal recomendación se extiende a casos donde existan bajos o casi nulos niveles de ligamiento como sucede en colecciones de germoplasma de especies autógamas genotipadas con cantidades relativamente bajas de marcadores. El objetivo de este trabajo fue comparar el desempeño, a nivel de las tasas de falsos positivos, de tres modelos biométricos de mapeo asociativo bajo escenarios de baja estructuración genética y sin el uso de alta densidad de marcadores. Los modelos comparados fueron: uno que no incluye corrección por estructura genética (M1) y dos que consideran componentes principales (CPs) para representar estructura. De éstos, uno incorpora la estructura a través de CPs en un modelo lineal de efectos fijos (M2) y otro trata a la CPs como componentes aleatorias en un modelo lineal mixto (M3). La comparación se realizó sobre bases de datos reales y simulados de cebada.
MATERIALES Y MÉTODOS
Datos
Los datos analizados son de un programa de mejoramiento de cebada y comprenden 179 genotipos y 811 marcadores moleculares del tipo Diversity Array Technology (DArT®) (Comadran et al., 2009). Los datos simulados consideraron 2.100 marcadores moleculares y la presencia de estructura genética dada por la existencia de tres sub-poblaciones. Para la simulación se usó el software Easypop (Balloux, 2001) con la siguiente asignación de parámetros: sub-poblaciones sin cruzamientos aleatorios entre ellas, proporción clonal igual a cero, coeficientes de autofecundación igual a uno, modelo de migración de islas jerárquico, proporción de migración dentro de grupos igual a cero y proporción de migración entre grupos 0,1 y 0,3; tasa de recombinación entre loci de 0,03 y tasa de mutación igual a cero. Sobre los genotipos simulados se seleccionaron aleatoriamente diez marcadores de la matriz genética y en base a sus posiciones se generaron fenotipos distribuidos normalmente . La simulación implementada permitió conocer la ubicación de los marcadores que influyen en la expresión del fenotipo. Los marcadores seleccionados que se encontraban dentro en una ventana de 10 cM, fueron interpretados como QTL. Otros trabajos en cebada han usado la misma amplitud de ventana en la identificación de QTL (Comadran et al., 2009; von Zitzewitz et al., 2011). Cada simulación fue repetida 30 veces.
Modelos de mapeo asociativo
Se emplearon tres modelos de mapeo asociativo para evaluar el efecto del marcador sobre el fenotipo, M1: sin corregir por estructura genética poblacional y otros dos corrigiendo por estructura genética en las siguientes aproximaciones, M2: que utilizó como covariables los ejes significativos según TW de un ACP en un modelo lineal de efectos fijos y M3 que utilizó los mismos CPs como covariables en la porción aleatoria de un modelo lineal mixto:
donde es el vector de valores fenotípicos, es la matriz de datos de los marcadores moleculares, es el vector de efectos alélicos a estimar, es la matriz de estructura genética (componentes principales), es el vector de efectos de la estructura poblacional y es el vector de términos de error aleatorio.
Cuando se contrastaron las hipótesis los valores p fueron corregidos por multiplicidad según la propuesta de Benjamini y Hochberg (1995). El desempeño de los modelos se evaluó usando como criterio la proporción de falsos positivos (FP) y la proporción de falsos negativos (FN) obtenidas a través del conteo de asociaciones falsas detectadas como significativas (FP) o asociaciones no detectadas (FN) en cada simulación y promediando luego sobre las 30 simulaciones realizadas. Los análisis fueron realizados mediante Info-Gen (Balzarini y Di Rienzo, 2004) y su interfaz con R (R Development Core Team, 2013). El código para el ajuste de estos modelos de mapeo asociativo se presenta en Info-Gen, el cual invoca desde la interfaz mencionada los scripts desarrollados por Gutiérrez (2011).
RESULTADOS
Tanto con los datos reales como con los simulados los niveles de correlación entre marcadores fueron generalmente bajos. En la Figura 1 se presenta el heat map de los datos genéticos reales. En este gráfico se colorea cada asociación entre un par de marcadores según la magnitud de la misma. Los puntos más calientes (rojos) no son muchos y no se observa un patrón de asociación, sólo algunas correlaciones (0,26%) fueron mayores a 0,6.

Figura 1. Heat map de medidas de desequilibrio de ligamiento entre pares de marcadores (811 DArT).
El ACP realizado sobre de datos de marcadores, sugiere que ésta puede identificarse con 17 combinaciones lineales (CPs). En la Figura 2a se muestra la proporción de varianza explicada por cada CP. El estadístico de Tracy-Widom (TW) retuvo estos primeros 17 ejes como estadísticamente significativos; las cinco primeras componentes resumían el 40% de la variabilidad total. Para los datos simulados, TW requirió aún más ejes para resumir la estructura (41 ejes), siendo esa cantidad la necesaria para explicar un 60% de la variabilidad genética (Figura 2b). Estos resultados del estudio de la estructura genética, sugieren que la divergencia genética entre las subpoblaciones de datos reales es mayor que en la de datos simulados donde el estadístico Fst tuvo un valor relativamente bajo (0,01).
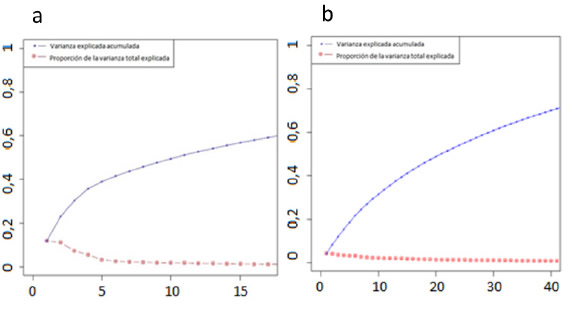
Figura 2a y 2b. Proporción total de varianza explicada por los ejes significativos según el estadístico de Tracy- Widom y varianza explicada acumulada a la izquierda para los datos reales y a la derecha para una de las realizaciones de los datos simulados.
La variabilidad fenotípica encontrada en los datos reales de cebada representó un 11% respecto a la media del rendimiento para los datos reales. Se detectaron marcadores significativos usando los valores máximos de - log10 (valor p) donde valor p es el nivel de significancia real asociado a la prueba de hipótesis de asociación nula entre un marcador y el carácter fenotípico. El valor umbral fue de tres, es decir que aquellos marcadores donde la transformación logarítmica es superior a tres son identificados como estadísticamente significativos o ligados a un QTL.
Para los datos reales, el modelo sin corrección por estructura y el modelo M2 mostraron valores máximos del estadístico - log10 (valor p) mayores a los observados en el modelo M3. Los valores p asociados a las pruebas de hipótesis bajo M3 fueron mayores, entiéndase con menor significancia. Al asignar un valor umbral de tres, es decir identificando como significativos aquellas asociaciones con un valor P menor a 0,001, ningún QTL fue detectado en la base de datos reales, mientras que para M1 y M2 la cantidad de marcadores significativos para el mismo valor umbral aumentó de manera considerable. Estos resultados muestran que el modelo M3 es más conservador que los otros dos, presentando menor tasa de FP.
En la Figura 3 se presenta la significancia de cada marcador analizado bajo los diferentes modelos en datos reales (arriba) y en datos simulados (abajo); los valores altos (mayores al umbral propuesto) sugieren la presencia de un potencial QTL. Los resultados sugieren que la inclusión de la estructura a través de las componentes principales en el modelo como efecto aleatorio produjo mayor control sobre la tasa de falsos positivos, i.e. se detectaron menor cantidad de FP. El M3 indicó que no existían QTL para el rendimiento.

Figura 3. Perfil de significancias para tres modelos de mapeo asociativo en datos reales (arriba) y en datos simulados (abajo), en el eje de las abcisas se representan los marcadores y en las ordenadas el estadístico
En los datos simulados se observó también una reducción del estadístico - log10 (valor p) en el modelo con CPs aleatorias. El modelo que usó las CPs como efectos fijos incrementó los valores de este estadístico y, consecuentemente, la tasa de falsos positivos, incluso más que aquel sin corrección por estructura.
En la Tabla 1 se muestra la proporción de falsos positivos (FP) y falsos negativos (FN) para los tres modelos evaluados. Se observa que la corrección por estructura incluyendo CPs como efecto aleatorio produce una disminución de las tasas de FP.
Tabla 1. Tasa de falsos positivos y de falsos negativos para tres modelos de mapeo asociativo.

M1: sin corrección por estructura, M2: con corrección mediante CPs como covariables de efectos fijos y M3: con corrección mediante CPs como covariables de efectos aleatorios.
Para la comparación de modelos se usó también la función de distribución acumulada de los valores P resultantes de las pruebas de hipótesis sin corrección por multiplicidad. En la Figura 4 se muestra la función de distribución empírica para los valores P de cada modelo para datos reales (izquierda) y simulados (derecha). Tanto para los datos reales como para los simulados, basados en la distribución de los valores P, el modelo sin corrección por estructura y el que introduce las componentes principales como efecto fijo, no se desempeñaron bien. Para ambos se observa una distribución asimétrica hacia valores P pequeños, es decir, mayor significancia de la esperada (i.e. mayor tasa de FP).

Figura 4. Función de distribución acumulada de los valores P para datos reales (a) y simulados (b).
DISCUSIÓN
El mapeo asociativo o mapeo por LD se usa para detectar asociaciones entre caracteres cuantitativos y marcadores moleculares (Melchinger et al., 1998). A diferencia del análisis de QTL clásico el cual requiere la generación de poblaciones de mapeo, tales como F2, retrocruzas y/o dobles haploides (Bonamico et al., 2010), el mapeo por LD puede llevarse a cabo con poblaciones de líneas de fácil obtención por ejemplo colecciones de germoplasma. Sin embargo, el número limitado de eventos de recombinación que pueden acumular estas poblaciones de mapeo, podría resultar en una baja resolución para la detección de QTL. El número de alelos por locus en la población de mapeo puede ser chico en relación a la diversidad genética disponible para las especies y, por tanto, el contenido de información sería bajo. Una forma de tratar este problema es aumentar el número de loci en estudio. Mientras más denso es el genotipado, más desequilibrio de ligamiento o correlación entre los marcadores se espera encontrar.
Además, el LD también está asociado a la historia de recombinación de las poblaciones. En autógamas, con poblaciones genotipadas por menos de 1.000 marcadores, el LD puede no ser alto y este hecho puede afectar la detección de marcadores significativos o ligados a los loci de interés. En el conjunto de datos de cebada usado en este trabajo, el LD fue bajo (r2<0,20). Kraakman et al. (2006) estudiaron el LD en poblaciones de cebada y concluyeron que puede existir LD hasta intervalos de 10 cM entre marcadores, pero también con valores bajos. Francia et al. (2004) y von Zitzewitz et al. (2011) en el mismo cultivo informaron QTL pertenecientes a dos grupos de LD en un mismo cromosoma.
Respecto a los modelos de mapeo asociativo en autógamas, Gutiérrez et al. (2011) y Wang et al. (2012) probaron métodos de corrección de la estructura poblacional, incluyendo entre ellos no solo al modelo con CPs aleatorias sino también al modelo que incorpora como covariables a la matriz de salida del software STRUCTURE (Pritchard et al., 2000). Los resultados mostraron comportamientos similares entre estas dos últimas estrategias. Los autores destacaron que la implementación del control por estructura con análisis de componentes principales fue significativamente más eficiente en tiempo computacional. Estos resultados coinciden con los encontrados en el presente estudio, ya que el modelo con las CPs aleatorias fue el único que no produjo QTL falsos. Existe información previa, con las mismas líneas de cebada usadas en este trabajo que indican la ausencia de QTL para rendimiento (Comadran et al., 2009).
En contextos de bajo LD y escasa estructuración genética, la incorporación de las CPs como efecto fijo puede producir una sobre-parametrización del modelo que conduce a un incremento en la tasa de FP. En nuestro estudio, al igual que lo informado por Malosetti et al. (2007) y Gutiérrez et al. (2011), el modelo con CP de efecto fijo y el modelo sin corrección por estructura produjeron un gran número de falsos positivos o asociaciones espurias. Este resultado debería ser frecuente en situaciones donde la estructura poblacional es de poca magnitud y el número de componentes necesario para resumirla resulta alto. En tal contexto, el modelo lineal a estimar usa una cantidad alta de parámetros para describir poca variabilidad. Cappa et al. (2013) al estudiar el comportamiento de diversos modelos de mapeo, propuestos por Yu et al. (2006) en poblaciones de eucalipto en Argentina y Uruguay, concluyeron que para cuatro de los seis caracteres estudiados, el modelo con las CPs aleatorias fue el mejor. La incorporación de las CPs como covariables de efectos aleatorios para contemplar la estructura poblacional, disminuyó, en nuestro trabajo, la tasa de falsos positivos.
ANEXO
Nota: “yield” es la variable respuesta para nuestros análisis
#Carga de archivos
qtl.data<-load.data(P.file=”QAssociation_pheno.txt”,G.file=”QAssociation_geno.txt”, map.file=”QAssociation_map.txt”, cross=”am”, heterozygotes=”FALSE”)
#Estudio de la estructura de población
pca<-pca.analysis(file=qtl.data, p.val=0.05)
#Estudio de la caída de LD dentro de los cromosomas
LD.plots(file=qtl.data, structure=”FALSE”, heterocigotes=”TRUE”)
#Asociaciones marcadores-caracteres utilizando diferentes modelos.
#1. Con las CPs aleatorias
(pcaR.am<-am(file=qtl.data,method=”mixed.random”,provide.K=FALSE,covariates= pca$scores, trait=”yield”, threshold=”FDR”, p=0.05, out.file=”AM fixed PCAmodel”))$selected
#2. Con las CPs fijas
(pca.am<-am(file=qtl.data, method=”fixed”, provide.K=FALSE, covariates=pca$scores, trait=”yield”,threshold=”FDR”, p=0.05,out.file=”AM fixed PCAmodel”))$selected
#3. Modelo sin correction por estructura
(naive.am<-am(file=qtl.data, method=”naive”, provide.K=FALSE, covariates=FALSE, trait=”yield”, threshold=0.05, p=0.05, out.file=”AM naïve model”))$selected
#Grafica los resultados para cada modelo, se presenta sólo un caso
p.file<-pcaR.am$p.val
xyplot(-log10(p.file[,3])~p.file[,2]|factor(p.file[,1]),type=”h”,layout=c(length(unique (p.file[,1])),1),col=”red”,xlab=”Posición de Cromosoma”,ylab=”-log10(P)”,main= “Mapeo Asociativo con CPs aleatorias”,scales=list(x=”free”),ylim=c(0,(max(-log10(p.file[,3]))+0.5)))
AGRADECIMIENTOS
Los autores agradecen a los revisores por sus valiosas sugerencias. El presente trabajo es parte del trabajo de tesis de Andrea Peña Malavera para el cumplimiento de los requisitos del Doctorado en Ciencias de la Ingeniería de la Universidad Nacional de Córdoba y del Programa de becas de posgrado del Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET).
BIBLIOGRAFÍA
1. Aranzana M.J., Kim S., Zhao K., Bakker E., Horton M., Jakob K., Lister C., Molitor J., Shindo C., Tang C., Toomajian C., Traw B., Zheng H., Bergelson J., Dean C., Marjoram P., Nordborg M. (2005) Genome-wide association mapping in arabidopsis identifies previously known flowering time and pathogen resistance genes. PLoS Genetics 1(5): e60. [ Links ]
2. Balloux F. (2001) EASYPOP (Version 1.7): A computer program for population genetics simulations. Journal of Heredity 92 (3): 301-302. [ Links ]
3. Balzarini M., Di Rienzo J. (2004) Info-Gen Córdoba: Universidad Nacional de Cordoba, Argentina. [ Links ]
4. Benjamini Y., Hochberg Y. (1995) Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B, 57: 289-300. [ Links ]
5. Bonamico N., Balzarini M., Arroyo A., Ibañez M., Díaz D., Salerno J., Di Renzo M. (2010) Association between microsatellites and resistance to Mal de Río Cuarto in maize by discriminant analysis. Phyton 79: 8. [ Links ]
6. Breseghello F., Sorrells M.E. (2006) Association mapping of kernel size and milling quality in Wheat (Triticum aestivum L.) cultivars. Genetics 172 (2): 1165-1177. [ Links ]
7. Cappa E.P., El-Kassaby Y.A., Garcia M.N., Acuña C., Borralho N.M.G., Grattapaglia D., Marcucci Poltri S.N. (2013) Impacts of population structure and analytical models in genome-wide association studies of complex traits in forest trees: A case study in eucalyptus globulus. PLoS ONE 8 (11): e81267. [ Links ]
8. Comadran J., Thomas W.T.B., Eeuwijk F.Á., Ceccarelli S., Grando S., Stanca A.M., Pecchioni N., Akar T., Al-Yassin A., Benbelkacem A., Ouabbou H., Bort J., Romagosa I., Hackett C.A., Russell J.R. (2009) Patterns of genetic diversity and linkage disequilibrium in a highly structured Hordeum vulgare association-mapping population for the Mediterranean basin. Theoretical and Applied Genetics 119 (1): 175-187. [ Links ]
9. Corder E., Saunders A.M., Strittmatter W.J., Schmechel D.E., Gaskell P.C., Rimmler J.B., Locke P.A., Conneally P.M., Schmader K.E., Small G.W., Roses A.D., Haines J.L., Pericak-vance M.A. (1994) Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer's disease. Nature Genetics 7: 4. [ Links ]
10. D'hoop B., Paulo M., Mank R., Eck H., Eeuwijk F. (2008) Association mapping of quality traits in potato (Solanum tuberosum L.). Euphytica 161 (1-2): 47-60. [ Links ]
11. Falconer D.S., Mackay T.F.C. (1996) Introduction to quantitative genetics. Harlow, UK. [ Links ]
12. Francia E., Rizza F., Cattivelli L., Stanca A.M., Galiba G., Tóth B., Hayes P.M., Skinner J.S., Pecchioni N. (2004) Two loci on chromosome 5H determine low-temperature tolerance in a 'Nure' (winter) x 'Tremois' (spring) barley map. Theoretical and Applied Genetics 108 (4): 670-680.
13. Gutierréz L. (2011) Mapeo Asociativo. Material en formato de documento portátil (pdf) del curso de posgrado de la Maestría en Ciencias Agrárias; utilización de herramientas genómicas en vegetales: análisis de QTL. UdelaR, Facultad de Agronomía. EEMAC Paysandú [ Links ].
14. Gutiérrez L., Cuesta-Marcos A., Castro A.J., von Zitzewitz J., Schmitt M., Hayes P.M. (2011) Association mapping of malting quality quantitative trait loci in winter barley: positive signals from small germplasm arrays. Plant Genome 4 (3): 256-272. [ Links ]
15. Hottelling H. (1936) Relations between two sets of variables. Biometrika 28: 321-377. [ Links ]
16. Kraakman A.T.W., Martínez F., Mussiraliev B., Eeuwijk F.A., Niks R.E. (2006) Linkage disequilibrium mapping of morphological, resistance, and other agronomically relevant traits in modern spring barley cultivars. Molecular Breeding 17 (1): 41-58. [ Links ]
17. Lynch M., Walsh B. (1998) Genetics and analysis of quantitative traits. Massachusetts, USA. [ Links ]
18. Malosetti M., van der Linden C., Vosman B., van Eeuwijk F. (2007) A mixed-model approach to association mapping using pedigree information with an illustration of resistance to phytophthora infestans in potato. Genetics 175 (2): 879-889. [ Links ]
19. Melchinger A.E., Utz H.F., Schön C.C. (1998) Quantitative Trait Locus (QTL) mapping using different testers and independent population samples in maize reveals low power of QTL detection and large bias in estimates of QTL effects. Genetics 149 (1): 383-403. [ Links ]
20. Muñoz-Amatriaín M., Cuesta-Marcos A., Endelman J.B., Comadran J., Bonman J.M., Bockelman H.E., Chao S., Russell J., Waugh R., Hayes P.M., Muehlbauer G.J. (2014) The USDA Barley core collection: genetic diversity, population structure, and potential for genome-wide association studies. PLoS ONE 9 (4): e94688. [ Links ]
21. Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics 38 (8): 5. [ Links ]
22. Pritchard J., Stephens M., Donnelly P. (2000) Inference of population structure using multilocus genotype data. Genetics 155: 945-959. [ Links ]
23. R Development Core Team (2013) R: A language and environment for statistical computing. R Fundation for Statistical Computing, Vienna, Austria. URL http://www.R-project.org. [ Links ]
24. Remington D., Thornsberry J., Matsuoka Y., Wilson L., Whitt S., Doebley J., Kresovich S., Goodman M., Buckler E. (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proceeding of National Academy of Science USA 98 (20): 6. [ Links ]
25. Searle S.R., Casella G., McCulloch C.E. (2008) Maximum likelihood (ML) and restricted maximum likelihood (REML), in Variance Components. John Wiley & Sons, Inc. pp. 232-257. [ Links ]
26. Stich B., Melchinger A., Heckenberger M., Möhring J., Schechert A., Piepho H.P. (2008) Association mapping in multiple segregating populations of sugar beet (Beta vulgaris L.). Theoretical and Applied Genetics 117 (7): 1167-1179. [ Links ]
27. Thornsberry J., Goodman M., Doebley J., Kresovich S., Nielsen D., Buckler E. (2001) Dwarf8 polymorphisms associate with variation in flowering time. Nature Genetics 28: 3. [ Links ]
28. Tracy C.A., Widom H. (1994) Level-spacing distributions and the airy kernel. Communications in Mathematical Physics 159 (1): 23. [ Links ]
29. von Zitzewitz J., Cuesta-Marcos A., Condon F., Castro A.J., Chao S., Corey A., Filichkin T., Fisk S.P., Gutierrez L., Haggard K., Karsai I., Muehlbauer G.J., Smith K.P., Veisz O., Hayes P.M. (2011) The genetics of winterhardiness in barley: Perspectives from genome-wide association mapping. Plant Genome 4 (1): 76-91. [ Links ]
30. Wang M., Jiang N., Jia T., Leach L., Cockram J., Waugh R., Ramsay L., Thomas B., Luo Z. (2012) Genome-wide association mapping of agronomic and morphologic traits in highly structured populations of barley cultivars. Theoretical and Applied Genetics, pp.: 1-14. [ Links ]
31. Yu J., Buckler E.S. (2006) Genetic association mapping and genome organization of maize. Current Opinion in Biotechnology 17 (2): 155-160. [ Links ]
32. Yu J., Pressoir G., Briggs W.H., Vroh Bi I., Yamasaki M., Doebley J.F., McMullen M. D., Gaut B.S., Nielsen D.M., Holland J.B., Kresovich S., Buckler E.S. (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nature Genetics 38 (2): 203-208. [ Links ]
33. Zhang Z., Ersoz E., Lai C.Q., Todhunter R.J., Tiwari H.K., Gore M.A., Bradbury P.J., Yu J., Arnett D.K., Ordovas J.M., Buckler E.S. (2010) Mixed linear model approach adapted for genome-wide association studies. Nature Genetics 42 (4): 355-360. [ Links ]
34. Zhu C., Gore M., Buckler E., Yu J. (2008) Status and prospects of association mapping in plants. The Plant Genome 1 (1): 16. [ Links ]

