










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de la Asociación Argentina de Ortopedia y Traumatología
versión On-line ISSN 1852-7434
Rev. Asoc. Argent. Ortop. Traumatol. vol.83 no.1 Ciudad Autónoma de Buenos Aires mar. 2018
PRESENTACIÓN DE CASOS
Síndrome de Grebe.
Reporte de un caso
Jessica A. Suárez Zarrate,* Ricardo Arias Arguello,** Sebastián Rodríguez Serna**
*Fundación Universitaria Sanitas, Bogotá, Colombia
**Clínica Universitaria Colombia, Bogotá, Colombia
Dra. Jessica A. Suárez Zarrate • jessi_k10@hotmail.com
Conflicto de intereses: Los autores no declaran conflictos de intereses.
Recibido el 8-12-2016.
Aceptado luego de la evaluación el 1-8-2017
Resumen
La condrodisplasia de Grebe es un trastorno raro autosómico recesivo que pertenece al grupo de las osteocondrodisplasias. Clínicamente se caracteriza por un severo dismorfismo con una marcada micromelia y deformidad de las extremidades inferiores y superiores. Conocer este tipo de síndrome orienta a dar mejores diagnósticos y permite el diagnóstico diferencial con patologías más comunes, como la acondroplasia.
Se presenta una paciente de 35 años con diagnóstico de síndrome de Grebe desde los 10 años. El síndrome de Grebe tiene una muy baja incidencia; por este motivo, es poco conocido por el cuerpo médico en general y aun menos para los ortopedistas, quienes serán los encargados de tratar a estos pacientes.
Palabras clave: Síndrome de Grebe; Osteocondrodisplasia; Displasia; Acromesomélico.
Nivel de Evidencia: IV
Abstract
Grebe syndrome. Case report
Grebe syndrome is a rare autosomal recessive disorder that belongs to the group of osteochondrodysplasias. Clinically, it is characterized by severe dysmorphism, marked micromelia and deformities of the lower and upper limbs. Recognition of this syndrome allows to give better diagnoses and to establish a differential diagnosis with more common pathologies, such as achondroplasia.
We present a 35-year-old woman with diagnosis of Grebe syndrome at the age of 10. Grebe syndrome has a very low incidence; therefore, it is unknown by general physicians and still less by orthopedic surgeons, who will treat these patients.
Key words: Grebe syndrome; Osteochondrodysplasia; Dysplasia; Acromesomelic.
Level of Evidence: IV
Introducción
La condrodisplasia de tipo Grebe es un trastorno raro autosómico recesivo que pertenece al grupo de las osteocondrodisplasias; se han publicado 25 casos. El gen responsable ya está identificado y codifica para la proteína derivada del cartílago morfogénico 1 (CDMP-1) y está localizado en el cromosoma 20q11.2.1 Desde el punto de vista clínico, se caracteriza por un severo dismorfismo con marcada micromelia y deformidad de las extremidades inferiores y superiores, con una gravedad típica que va aumentando de proximal a distal. Los dedos se reducen a apéndices globulares (puente de tejido blando), no hay aparente desarrollo del pulgar ni aparente articulación de los dedos de los pies (sin movimiento) y limitación de la movilidad en codos, muñecas, rodillas, tobillos y dedos. Son comunes las luxaciones en las articulaciones proximales de las extremidades. El esqueleto axial, la cabeza y el aspecto facial tienen características normales. Los pacientes tienen talla baja, miden aproximadamente 100 cm.1 Radiológicamente se caracteriza por deformidad y acortamiento de huesos largos, fusión de huesos del carpo y del tarso, displasia severa de metatarsianos y metacarpianos, y en ocasiones, ausencia de las falanges medias y proximales.2
Conocer este tipo de síndrome orienta a dar un mejor diagnóstico, a descartar otras patologías similares, como la acondroplasia y ayuda a aclarar qué tipo de manifestaciones clínicas y hallazgos radiológicos presentan estos pacientes.
Caso clínico
Mujer de 35 años, peso 40 kg, talla 100 cm, con diagnóstico de síndrome de Grebe desde los 10 años. Una hermana y dos tías maternas en segundo grado padecen el mismo síndrome (Figura 1).
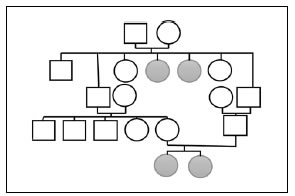
Figura 1. Familiograma de la paciente que muestra en dos generaciones, cuatro afectados.
En 2009, consulta por dolor en el codo provocado por la manipulación del mouse en el trabajo y se le diagnostica por imágenes luxación de la cúpula radial derecha. Se decide realizar una artroplastia de resección de la cúpula radial derecha. A los cinco meses del procedimiento, consulta por dolor en el codo (disestesias), limitación funcional y sensación subjetiva de paresia en el miembro superior derecho. Se sospecha una neuroapraxia del interóseo posterior. En el examen físico, se observan arcos de movilidad y fuerza conservados, con alodinia en el codo y el antebrazo derechos, por lo cual se indican valoración por clínica del dolor, fisioterapia y parches con lidocaína. Se realiza electromiografía, cuyo resultado es normal. Algunas manifestaciones clínicas del síndrome de Grebe son enanismo severo (100 cm), micromelia y deformidad en miembros superiores e inferiores, con diferentes grados de severidad. La apariencia facial, la inteligencia y el esqueleto axial son normales, y los miembros superiores e inferiores tienen segmentos proximales cortos y el medio aún más corto. Los arcos de movilidad son limitados en codos, muñecas, rodillas, tobillos y dedos. Hay anomalías graves de manos y pies, con dedos que se reducen a apéndices globulares (puente de tejido blando), no hay aparente desarrollo del pulgar, ni articulación aparente en los dedos de los pies (sin movimiento) (Figura 2).
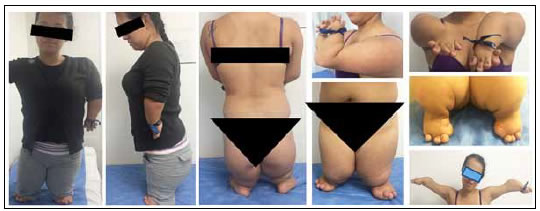
Figura 2. Paciente de 35 años con síndrome de Grebe.
Paciente completamente funcional, ha finalizado sus estudios universitarios y trabaja sin ningún inconveniente, totalmente independiente en sus actividades diarias, inclusive el aseo personal. Solo refiere dificultad para acceder al transporte público (colectivos) y conseguir calzado cómodo. En las radiografías, se detectó un esqueleto axial normal (Figura 3), acortamiento de huesos largos sin alteración de su morfología, fusión de huesos del carpo y del tarso, displasia severa de metatarsianos y metacarpianos (Figura 4) y luxación de la cúpula radial bilateral (Figura 5).
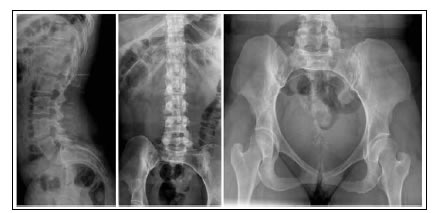
Figura 3. Radiografías de columna lumbosacra y caderas con displasia acetabular izquierda.
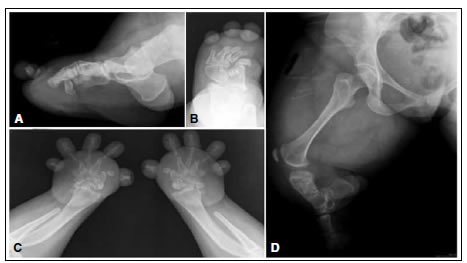
Figura 4. A. Radiografía de fémur y húmero que muestra acortamiento sin alteración de la morfología. B. Acortamiento con alteración de la morfología del radio y el cúbito, y la tibia y el peroné. C. Radiografía de mano y antebrazo bilateral. Ausencia parcial del 1º. y 5º. metacarpianos, ausencia de las falanges medias y fusión del hueso trapecio con el trapezoide, y del hueso grande con el ganchoso de la mano izquierda. D. Radiografía de pie derecho lateral. Ausencia parcial del 1er. metatarsiano, ausencia de falanges medias y fusión del hueso escafoides con la 1ª. cuña y del hueso cuboides con la 2ª. y 3ª. cuña.
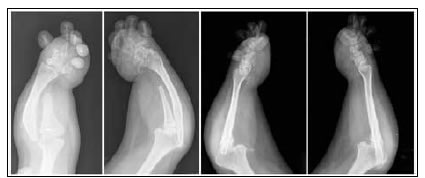
Figura 5. Radiografías de antebrazo y codo, y lateral derecha. Acortamiento de huesos del antebrazo con luxación de la cúpula radial bilateral.
Discusión
Las características fenotípicas de la paciente coinciden con el diagnóstico de condrodisplasia de tipo Grebe. Se han notificado casos de muerte neonatal, pero esta paciente ya ha llegado a la adultez sin ninguna complicación conocida. La micromelia severa es una anomalía frecuente en pacientes con atelosteogénesis de tipo 2 (condrodisplasia letal); otras características son las anormalidades esqueléticas, como hendiduras de la corona de los cuerpos vertebrales, anomalías epifisarias y metafisarias, pie equino varo, pulgares en gatillo.3 Las anomalías esqueléticas, en general, son totalmente diferentes de las de nuestra paciente.
Otras formas de acortamiento mesomélico, como el síndrome de Grebe, se caracterizan por displasia acromesomélica, falanges muy cortas con dedos en botón muy similares. Los hallazgos radiológicos incluyen ausencia de peroné, hipoplasia o displasia de antebrazos, displasia o ausencia de las falanges medias y proximales y, en algunos pacientes, la fusión del carpo.4
En 1981, Langer y cols. describen el síndrome de Grebe como una entidad clínica similar a los síndromes de Du Pan y Hunter-Thompson. Los fenotipos de estos síndromes son distintos, por lo cual se considera como una entidad totalmente diferente. Los pacientes con condrodisplasia de tipo Grebe tienen complejas anomalías en el esqueleto periférico, que se manifiestan con acortamiento de huesos largos con mayor compromiso a nivel distal, especialmente en pies y manos.5
Conclusiones
El síndrome de Grebe tiene una muy baja incidencia; por este motivo, es poco conocido por el cuerpo médico en general y aun menos para los ortopedistas, quienes por tener contacto con patologías musculoesqueléticas serán los encargados de manejar a estos pacientes.
Conocer este síndrome permite establecer el diagnóstico diferencial con patologías más comunes, como la acondroplasia, y reconocer qué manifestaciones clínicas y hallazgos radiológicos presentan estos pacientes como, en este caso, una luxación bilateral de la cúpula radial. De esta manera, se puede enfocar a este grupo de pacientes más objetivamente y lograr un manejo integral de sus cuadros.
1. Faivre L, Coermier Daire V. Acromesomelic dysplasia, Grebe type. Orphanet Encyclopedia http://www.orpha.net/data/patho/GB/uk-grebe05.pdf. [ Links ]
2. Costa T, Ramsby G, Cassia F, Peters KR, Soares J, Correa J, et al. Grebe syndrome: clinical and radiographic findings in affected individuals and heterozygous carriers. Am J Med Genet 1998;75(5):523-9. [ Links ]
3. Baraitser M, Winter RM. London Dysmorphology Database, London Medical Databases, Oxford: Oxford University Press. [ Links ]
4. Du Pan CM. Absence congenitale du perone sans deformation du tibia: Curieuses deformations congenitales des mains. Rev Orthop 1924;11:227-34. [ Links ]
5. Langer LO Jr, Cervenka J, Camargo M. A severe autosomal recessive acromesomelic dysplasia, the Hunter-Thompson type, and comparison with the Grebe type. Hum Genet 1989;81(4):323-8. [ Links ]

