










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista argentina de radiología
versión On-line ISSN 1852-9992
Rev. argent. radiol. vol.74 no.4 Ciudad Autónoma de Buenos Aires oct./dic. 2010
NEURORRADIOLOGÍA - REPORTE DE CASO
Síndrome de MELAS: TC y RM como herramienta diagnóstica no invasiva
Laura García, Romina Guglielmo, Lisandro Illa
Centro Médico Dr. Deragopyan. Amenábar 1457. (1425) C.A.B.A
Correspondencia: Laura C. García - laugarcia48@hotmail.com
Recibido: marzo de 2010, received: march 2010
Aceptado: julio de 2010; accepted: july 2010
®SAR-FAARDIT 2010
Resumen
Se presenta el caso de un paciente masculino, de 16 años, con hipoacusia bilateral, miopatías y alteración neurológica focal. Desde los 9 años, presenta deficiencia en el desarrollo muscular con episodios repetidos de intolerancia al ejercicio, así como miocarditis viral. Tras descartar etiología infecciosa se realizaron TC y RM, las cuales arrojaron datos positivos a nivel de los ganglios basales y del parénquima encefálico.
Los hallazgos radiológicos junto con la clínica del paciente sugirieron una encefalopatía de origen mitocondrial. El examen genético confirmó el diagnóstico de síndrome de MELAS (mitochondrial encephalomyopathy with lactic acidosis and stroke like events).
Palabras claves: Acidosis láctica; MELAS; Miopatía mitocondrial; RM; TC.
Abstract
MELAS syndrome: CT and MRI as a noninvasive diagnostic tool.
We report on a 16-year-old male with bilateral hypoacusia, myopathy and focal neurological disorder. Since 9-year-old, he showed muscle development deficiency with repeated episode of exercise in tolerance and viral myocarditis.
After ruling out an infectious etiology, CT and MRI were performed and yielded positive data about basal ganglia and brain parenchyma.
The patient's radiological and clinical findings suggested mitochondrial encephalopathy. Genetic testing confirmed the diagnosis of MELAS syndrome (lactic acidosis with mitochondrial encephalomyopathy and stroke-like events).
Keywords: C.T.; Lactic acidosis; MELAS; Mitochondrial myopathy; MRI.
INTRODUCCIÓN
El síndrome de MELAS (mitochondrial encephalomyopathy with lactic acidosis and stroke like events) es un desorden mitocondrial. Descripto, por primera vez, por Pavlakis en 1984 (1), se caracteriza por presentar encefalomiopatía, acidosis láctica y episodios semejantes a la apoplejía. Este síndrome está asociado a una mutación genética del ADN mitocondrial (ARNt transición A>G en la posición 3243) en el 80% de los casos aproximadamente y provoca un defecto en la síntesis proteica (2).
Las manifestaciones clínicas aparecen entre los 5 y 15 años de edad y, una vez presentados los síntomas, tiene un curso evolutivo rápido (3).
El diagnóstico se realiza mediante una biopsia muscular en la que se encuentra una lesión de fibras rojas (ragged red fibers), o bien con análisis genético en el que se pueden detectar mutaciones en el ARNt mitocondrial (4).
En la actualidad el uso de métodos de diagnóstico por imágenes como TC y RM (espectroscopía y difusión) son de gran utilidad en la aproximación diagnóstica de este síndrome.
PRESENTACIÓN DEL CASO
Paciente masculino de 16 años, con una estatura baja para la edad, que consultó por episodios de vómitos, cefaleas, síndrome cerebeloso, trastornos de la conciencia con episodios de desorientación y excitación psicomotriz con alteraciones visuales (diplopía).
Se realizó una RM en otro centro de salud con hallazgo presuntivo de encefalitis viral.
Se examinó el LCR (Líquido Céfalo-Raquídeo) a través de PCR (reacción en cadena de polimerasa) con resultado negativo para Herpes Virus.
A los 4 meses del primer episodio, presentó nuevamente vómitos y convulsiones generalizadas precedidas por fotopsias.
Se solicitó interconsulta con el servicio de Otorrinolaringología y se constató hipoacusia bilateral.
Se efectuó un nuevo examen de LCR, TC, RM, estudio de lactato sérico venoso y examen genético.
La TC evidenció la presencia de calcificaciones bilaterales y simétricas de los ganglios basales (distribución en banda) y una hipodensidad córtico-subcortical que no comprometía al territorio vascular de la arteria cerebral posterior derecha (Fig. 1).

Fig. 1. TC en corte axial sin contraste. Muestra calcificación de ganglios basales y compromiso vascular en territorio de arteria cerebral posterior derecha.
La RM en secuencias ponderadas en T1, T2 y FLAIR demostró dilatación moderada del sistema ventricular supratentorial, imágenes hiperintensas en las secuencias T2 y FLAIR a nivel córtico-subcortical de la convexidad de los lóbulos temporo-occipitales (Fig. 2 y 3) y, en ambos centros ovales (de características inespecíficas); hipointensidad de la sustancia gris y blancadel lóbulo temporal derecho; señal hiperintensa de los núcleos basales probablemente relacionada a la calcificación de los mismos; hiperintensidad simétrica y bilateral de ambos pulvinares (pulvinar sign) siendo el signo más evidente de astrocitosis (proceso irreversible que refleja una respuesta al daño sufrido por el SNC) (Fig. 4). La secuencia de difusión evidenció una restricción de la difusión a nivel córtico-subcortical de la región temporo-parietal derecha (Fig. 5). En la espectroscopía se registró un pico de lactato en la región que representaba restricción de la difusión (Fig. 6). La angio RM no mostró alteraciones (Fig. 7).

Fig. 2 (a y b). RM cerebro sin contraste cortes axiales secuencia ponderada T2. Áreas hiperintensas temporo-occipitales.
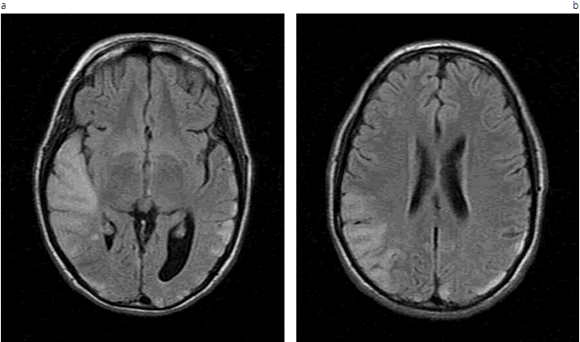
Fig. 3 (a, b). RM cerebro sin contraste, cortes axiales secuencias FLAIR. Hiperintensidad córtico-subcortical temporo-occipital.
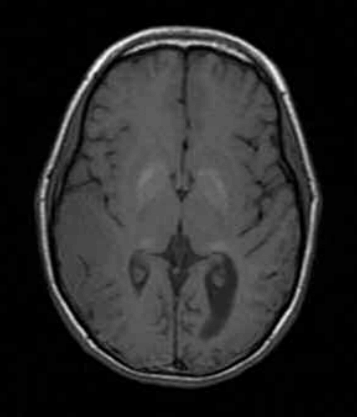
Fig. 4. RM de cerebro sin contraste, cortes axiales secuencias ponderadas T1. Vease la hiperintensidad bilateral y simétrica en ganglios basales y pulvinares (pulvinar sing).

Fig. 5 (a y b): RM de cerebro sin contraste. Difusión y ADC.

Fig. 6 (a y b). Espectroscopía. a) eco corto TE 36; b) eco largo TE 144. Pico de lactato (1.3 ppm) (ver círculos).

Fig. 7. AngioRM. Sin Alteraciones.
En el informe de laboratorio el ácido láctico en sangre venosa mostró valores elevados: 6 mmol/l (valor normal 2,2 mmol/l). También se encontró ácido láctico en el LCR.
Se realizó un examen genético que arrojó ARNt mitocondrial MP3243 mutación 3243 A>G.
DISCUSIÓN
Las enfermedades mitocondriales son un grupo heterogéneo de desórdenes causados por defectos en la producción de energía intracelular , que afectan a los tejidos con alto requerimiento energético (músculo y cerebro).
Clínicamente se manifiesta por debilidad muscular, intolerancia al ejercicio, oftalmoplejía, baja estatura y deterioro cognitivo lento y progresivo hasta llegar a la demencia. Suele asociarse a diabetes mellitus, sordera neurosensorial y miocardiopatía (5).
Su patogenia es provocada por una mutación a nivel ADN mitocondrial (ARNt posición 3243) que alteran la síntesis proteica de las enzimas del Ciclo de Krebs (COX1) y provocan un defecto en la cadena de transporte de electrones cuyo resultado es la academia láctica.
La RM demuestra ser superior a otros métodos diagnósticos al momento de efectuar el diagnóstico de la enfermedad mitocondrial.
Las secuencias más representativas son la espectroscopía (pico de lactato y disminución del pico de NAA) y la difusión (infartos parenquimatosos que no comprometen territorios vasculares y respeta sólo la corteza) (6).
El examen de LCR demuestra una elevación de los niveles de lactato, hallazgo frecuente en las enfermedades mitocondriales.
Los diagnósticos diferenciales del síndrome de MELAS a nivel clínico e imagenológico constituyen el síndrome de Pearson - Kearns Sayre Syndrome (KSS) y el síndrome de Leigh (4).
El KSS muestra una similar presentación clínica a excepción de la retinitis pigmentaria y los trastornos endocrinos.
Con respecto a su presentación en RM, difiere del síndrome de MELAS ya que en el KSS se observa una atrofia, que es más marcada a nivel del cerebelo. Las calcificaciones son de distribución difusa, a nivel del globo pálido y núcleo caudado, y la hiperintensidad en secuencias ponderadas en T2 son simétricas y comprometen al núcleo central del tálamo y el tegmentum del pedúnculo cerebral (3).
El síndrome de Leigh se asimila clínicamente, a excepción de la pérdida del control respiratorio en estadíos finales. Las imágenes en RM muestran un marcado compromiso putaminal, siendo hipointenso en secuencias ponderadas en T1 e hiperintenso en secuencias ponderadas en T2 y una presencia de severoretraso en la mielinización. La sustancia blanca de los lóbulos frontales y temporales se presenta isointensa con el LCR y el manto cortical muestra señal anómala.
Las imágenes de RM en el síndrome de MELAS no son patognomónicas, aunque cumplen un patrón característico con respecto a las enfermedades mitocondriales anteriormente descriptas, sobre todo por la presencia de infartos corticales temporo-occipitales sin respetar los territorios vasculares y la calcificación bilateral y simétrica de los ganglios basales.
Sin embargo, la hiperintensidad en secuencias ponderadas en T1, T2 y FLAIR en forma simétrica de ambos pulvinares (pulvinar sign) es orientativa de síndrome de MELAS, ya que este signo es específico para este síndrome y para otras 3 entidades como son la enfermedad de Creuzsteldf Jacob (variante), la de Wernicke Korsakoff (hipovitaminosis B1) y la de Fabry (7).
Los hallazgos descriptos en la literatura revisada coinciden con los hallados en este reporte de caso, con la diferencia de que la presencia del signo del pulvinar no ha sido demostrada en todos los casos reportados.
Cabe aclarar que, en el caso presentado, el diagnóstico se realizó a una edad avanzada, mientras en los casos hallados en la literatura el mismo se había realizado a una edad más temprana, ya que las manifestaciones clínicas aparecen en la infancia.
CONCLUSIÓN
Las enfermedades mitocondriales presentan hallazgos positivos en los estudios de imágenes. El compromiso se presenta a nivel de los ganglios basales, pulvinares (pulvinar sign) y mantos corticales.
En el síndrome de MELAS dichos hallazgos son específicos y muestran calcificación de los ganglios basales e infartos que no respetan los territorios vasculares.
Los hallazgos en espectroscopia presentan disminución del pico de NAA y aparición del pico de lactato no siendo específicos de esta enfermedad.
Consideramos que para arribar a un diagnóstico de certeza se tiene que abarcar el síndrome desde un enfoque multidisciplinario (clínica, bioquímica, neuroradiología y genética) siendo las imágenes una herramienta primordial para ello.
Agradecimientos
Nos es grato agradecer la supervisión y el apoyo académico del Dr. Eduardo Mondello y la confianza depositada por parte de los Dres. Jaime Rimoldi y Rolando Deragopyan.
Ala Dra. Verónica Alarcón por su incondicional ayuda en la recaudación de datos. Ya las Licenciadas Analía Martino y Anabel Cardarelli por la colaboración técnica y profesional que nos prestaron durante la realización de este trabajo.
1. Cano A, Romero AI, Bravo F, et al. Síndrome de Melas: hallazgos neurorradiológicos. Comunicación Breve. Radiología. 2002; 44 (2): 69-74. [ Links ]
2. Clark JM, Marks MP, Adalsteinsson E, et al. MELAS: Clinical and Patologic correlations with MRI, xenon/CT, and MR spectroscopy. Neurology. 1996; 46: 226-7. [ Links ]
3. Singh SK, Sarin D, Puliyel JM, et al. Melas Syndrome. Clinical Brief. Indian J Pediatr. 1999; 66 (4): 621-5. [ Links ]
4. Valanne L, Ketonen L, Majander A, et al. Neuroradiologic Findings in Children with mitocondrial disorders. AJNR Am J Neuroradiol.1998; 19(2): 369-77. [ Links ]
5. Abe K, Yoshimura H, Tanaka H, Fujita N, Hikita T, Sakoda S. Comparison of conventional and diffusion-weighted MRI and proton MR spectroscopy in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke like events. Neuroradiology. 2004; 46 (2):113-7. [ Links ]
6. Bianchi MC, Tosetti M, Battini R, et al. Proton MR spectroscopy of mitochondrial diseases: analysis of brain metabolic abnormalities and their possible diagnostic revelance. AJNR Am J Neuroradiol. 2003; 24 (10): 1958-66. [ Links ]
7. Young GS, Geschwind MD, Fischbein NJ, et al. Diffusionweighted and fluid-attenuated inversion recovery imaging in Creutzfeldt-Jakob disease: high sensitivity and specificity for diagnosis. AJNR Am J Neuroradiol.2005; 26 (6): 1551 - 62. [ Links ]

