










Servicios Personalizados
Revista

Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista argentina de radiología
versión On-line ISSN 1852-9992
Rev. argent. radiol. vol.80 no.2 Ciudad Autónoma de Buenos Aires jun. 2016
CARTAS CIENTÍFICAS
Bilioescroto como complicación rara de la coledocolitiasis: a propósito de un caso de biloma retroperitoneal
Bilioscrotum as a rare complication of choledocholithiasis: Presentation of a case of retroperitoneal biloma
Estimada Editora:
Las lesiones de la vía biliar pueden producir colecciones de bilis, llamadas bilomas, que suelen localizarse en el espacio peritoneal o subcapsular hepático. Sin embargo, la bilis también puede distribuirse excepcionalmente por el espacio retroperitoneal, simulando una patología pancreática, aórtica, renal o incluso testicular, si esta sustancia irritante alcanza el canal inguinal y llega a la bolsa escrotal1.
Los bilomas retroperitoneales se producen por lesiones de origen iatrogénico, traumático o menos comúnmente de forma espontánea en la vía biliar. La rotura espontánea sucede en general por hiperpresión secundaria a ectasia de contenido biliar (debida en su mayoría a cálculos localizados en la vía biliar extrahepática)2. Estas litiasis impiden el adecuado drenaje, por lo que dan lugar a un aumento del diámetro de la vía biliar, al punto que pueden llegar a generar necrosis de los ductos. Con menor frecuencia, la rotura espontánea es idiopática o secundaria a tumores, infartos o abscesos3,4.
Nuestro objetivo es comunicar un caso de biloma retroperitoneal con extensión de bilis al escroto, secundario a una lesión espontánea de la vía biliar por coledocolitiasis. El paciente, un varón de 68 años sin antecedentes de interés, fue evaluado en el servicio de urgencias por un dolor en la fosa ilíaca derecha, que se irradiaba al escroto (donde existía una tumoración de una semana de evolución). No se registró fiebre, ni signos de peritonismo o de obstrucción intestinal. Solo se destacó una leve ictericia.
En el laboratorio presentó hiperglucemia, un ligero aumento de la creatinina y de la fosfatasa alcalina, e hipertransaminasemia con bilirrubina de 6 mg/dl. Se le realizó una ecografía inguinal por sospecha de hernia incarcerada, en la que se descartó esta posibilidad, pero se observó el cordón espermático engrosado y distendido por líquido (fig. 1).

Figura 1 Corte ecográfico axial del cordón espermático en la región inguinal. Este se encuentra engrosado y distendido por líquido con septos y puntos ecogénicos. Además, se visualizan estructuras vasculares en el interior y grasa hiperecogénica (asterisco).
Dado que no se pudo arribar a un diagnóstico concluyente, posteriormente se llevó a cabo una tomografía computada (TC). Esta reveló una gran colección retroperitoneal, con extensión en los planos interfasciales y retromesentérico, que caudalmente comunicaba con la bolsa escrotal (fig. 2). Al hallazgo se sumaba la presencia de una leve dilatación de la vía biliar intra y extrahepática con una coledocolitiasis y colelitiasis. No existían fugas demostrables de contraste en el tracto genitourinario.

Figura 2 Tomografía computada sin contraste endovenoso. En el corte sagital oblicuo se aprecia una colección retroperitoneal de baja densidad (asterisco) que se extiende desde la región hepática posterior y pasa por los planos retromesentéricos e interfasciales hasta alcanzar la región inguinal.
Con vistas a precisar el diagnóstico, se practicó una punción ecoguiada de la colección retroperitoneal con aguja de calibre 18 G, utilizando un abordaje lumbar. Se obtuvieron 20 cc de muestra y en el análisis la presencia de bilirrubina confirmó la existencia de un biloma.
El paciente fue intervenido de urgencia para el drenaje quirúrgico de la colección retroperitoneal y luego se le realizó, de forma programada, una resonancia magnética (RM) con secuencia colangiográfica y una colangiopancreatografía retrógrada endoscópica (CPRE) con la extracción de la coledocolitiasis (figs. 3 y 4). La evolución clínica fue buena.

Figura 3 Resonancia magnética con secuencia colangiográfica en la que se observa una coledocolitiasis (flecha) que produce dilatación de la vía biliar.

Figura 4 Corte axial de resonancia magnética ponderada en T2 con saturación de grasa en la que se observa la vesícula retraída con gran litiasis ocupando prácticamente la totalidad de la luz del cuerpo y el fundus (flecha). Nótese la ocupación del espacio retroperitoneal por la bilis (asterisco).
La colangiorresonancia se erige como una técnica no invasiva excelente para la evaluación tanto de la vía biliar como del parénquima hepático, por lo que su rendimiento diagnóstico supera a la CPRE diagnóstica y a la colangiografía transhepática percutánea (CTP). Además, permite el uso de contrastes hepatoespecíficos basados en manganeso o gadolinio con excreción biliar, aunque su utilización no esté muy extendida.
Cuando no es posible la realización de una RM para determinar el punto de fuga y la causa subyacente del biloma, el estudio hepatobiliar por medicina nuclear o la colangiografía intravenosa por TC puede ayudar a localizar la lesión de la vía biliar, ya que esta última presenta una gran resolución para los pequeños ductos biliares más periféricos5. De todos modos, es difícil demostrar la fuga, si no es activa en el momento de la exploración4.
El biloma debe ser evacuado de forma quirúrgica o mediante abordaje percutáneo para prevenir la estasis del fluido y disminuir el riesgo de infección. En general, se prefiere el drenaje percutáneo del biloma en pacientes en los que el riesgo quirúrgico es alto.
Con respecto a las fugas biliares, algunas de ellas se autolimitan, pero en otras es necesario la descompresión de la vía biliar para reducir la presión dentro de los ductos. La CPRE constituye la técnica terapéutica de elección para la extracción del cálculo mediante papilotomía. No obstante, cuando la vía biliar no es accesible por vía retrógrada (p. ej. en reconstrucciones en Y de Roux), el drenaje biliar percutáneo transhepático o la exploración quirúrgica son necesarios4.
Saludan con distinguida consideración, Los autores
Confidencialidad de los datos
Los autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
I. Daimiel Naranjo*, D. Castaño Pardo y A. Martínez Arnaiz
Hospital Universitario 12 de Octubre, Madrid, España
* Autor para correspondencia.
Correo electrónico: idaimiel@hotmail.com (I. Daimiel Naranjo).
1. Brady RR, McAteer E, Weir CD. Biliscrotum and retroperitoneal biloma: spontaneous rupture of the biliary system presenting as an incarcerated inguinal hernia. Ulster Med J. 2006;75: 85 -7. [ Links ]
2. Rastogi R, Rastogi V. Case report: retroperitoneal biliary fluid collections secondary to common bile duct rupture - an unusual complication of choledocholithiasis in a child. Indian J Radiol Imaging. 2008;18:232 -5. [ Links ]
3. Lee JH, Suh JI. A case of infected biloma due to spontaneous intrahepatic biliary rupture. Korean J Intern Med. 2007;22:220 -4. [ Links ]
4. Kang SB, Han HS, Min SK, Lee HK. Nontraumatic perforation of the bile duct in adults. Arch Surg. 2004;139:1083 -7. [ Links ]
5. Wald C, Scholz FJ, Pinkus E, Wise RE, Flacke S. An update on biliary imaging. Surg Clin N Am. 2008;88:1195 -220. [ Links ]
Neumonía por Pneumocystis jiroveci como debut del síndrome de inmunodeficiencia adquirida (sida)
Pneumocystis jiroveci pneumonia as debut of acquired immune deficiency syndrome (AIDS)
Estimada Editora:
El Pneumocystis jiroveci (PJ) es un hongo atípico que produce neumonía fundamentalmente en pacientes inmunodeprimidos. Puede ser la manifestación inicial del síndrome de inmunodeficiencia adquirida (sida) en los portadores que desconocen su condición, en los que no tienen acceso a las terapias antirretrovirales altamente activas o en los que estas fracasan. Esto debe tenerse en cuenta a la hora de atender a pacientes con clínica respiratoria inespecífica y cuyo estado inmunológico es desconocido, ya que un tratamiento inadecuado del cuadro o su retraso pueden empeorar el pronóstico. Por este motivo, es importante conocer los hallazgos radiológicos que pueden hacernos sospechar esta patología1,2.
Presentamos el caso de una mujer fumadora, de 47 años de edad, que acudió al servicio de Urgencias por presentar disnea progresiva de varias semanas de evolución, asociada a pérdida de peso, febrícula, astenia y tos (que había empeorado en las últimas horas). En la auscultación se registraron crepitantes finos en la base derecha y una saturación de oxígeno del 86%. En la radiografía simple de tórax (fig. 1) se apreció un discreto aumento de la densidad parahiliar, que era bilateral y simétrica con un patrón en vidrio esmerilado, por lo que se decidió completar el estudio con una tomografía computada (TC). Esta confirmó la existencia de múltiples áreas parcheadas de densidad en vidrio esmerilado, bilaterales, fundamentalmente parahiliares y en los lóbulos superiores (fig. 2), además de múltiples lesiones quísticas bilaterales de distinta morfología, tamaño y grosor de pared (fig. 3). Dado que el único antecedente conocido era el tabaquismo, se planteó como primera posibilidad una bronquiolitis respiratoria con enfermedad intersticial o una neumonía intersticial descamativa, y con menor probabilidad una infección fúngica por PJ. Se inició tratamiento con corticoides, pero posteriormente se recibió el resultado positivo para el virus de la inmunodeficiencia humana (VIH). Como consecuencia, se reorientó el diagnóstico, instaurando una conducta terapéutica para el PJ. La evolución fue buena.
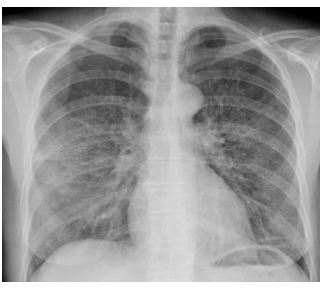
Figura 1 Radiografía simple de tórax donde se visualiza un discreto aumento de densidad, con patrón en vidrio esmerilado, bilateral y de predominio parahiliar.

Figura 2 Tomografía computada en plano axial donde se observa un aumento de densidad con un claro patrón en vidrio esmerilado, bilateral y de predominio parahiliar.

Figura 3 Tomografía computada en plano coronal donde se evidencian, en los lóbulos superiores, múltiples espacios aéreos quísticos bilaterales, de distintos tamaños, formas y grosor de pared (flechas). Algunos presentan en su interior opacidades que sugieren áreas de enfisema por patrón destructivo, en asociación con un patrón en vidrio esmerilado, bilateral, de predominio parahiliar y en los lóbulos superiores (círculo).
A pesar de que en este caso no se obtuvieron resultados positivos para el PJ en el lavado broncoalveolar ni en el esputo, el diagnóstico pudo realizarse en base a los hallazgos radiológicos, el estado inmunológico de la paciente y la mejoría tras el tratamiento médico. Por ello, para poder sospechar una neumonía por PJ en un paciente con un estado inmunitario desconocido, resulta importante conocer su clínica y radiología.
En los casos con VIH, la neumonía por PJ presenta un comienzo subagudo de hasta varias semanas, con síntomas como fiebre, tos seca, anorexia y pérdida de peso. La exploración resulta inespecífica y muestra desde crepitantes finos en las bases hasta taquipnea, taquicardia, hipoxia y cianosis. En la radiografía simple se pueden observar opacidades simétricas, difusas y bilaterales, preferentemente parahiliares, con apariencia de retículo nodulillar y patrón en vidrio deslustrado.
No obstante, los hallazgos radiológicos también pueden ser inespecíficos o incluso normales, por lo que en este último caso, si existe alta sospecha o se confirma que el paciente es VIH, se indica realizar una TC de alta resolución. Los hallazgos fundamentales serían: opacidades en vidrio esmerilado de predominio en lóbulos superiores, con preservación relativa de la periferia, asociadas a múltiples quistes pulmonares bilaterales, de forma, tamaño y grosor de pared variables, localizados en lóbulos superiores. A su vez, en estadios más avanzados pueden verse consolidaciones e incluso imágenes nodulares únicas o múltiples, aunque esto es mucho menos frecuente2,3.
Confidencialidad de los datos
Los autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
I. Belda Gonzáleza,*, D. Soliva Martínezb y V. Jiménez Castroc
a Servicio de Radiodiagnóstico, Hospital Virgen de la Luz, Cuenca, España
b Servicio de Radiodiagnóstico, Hospital de Alcañiz, Teruel, España
c Servicio de Neumología, Hospital Virgen de la Luz, Cuenca, España
* Autor para correspondencia: C/ Almería, n.◦ 8. CP: 23500. Jódar (Jaén). España. Tel.: +670288134.
Correo electrónico: isa_belda@yahoo.es (I. Belda González).
1. Cortés-Télles A, Juárez Hernández F, Peña Mirabal ES. Neumonía por Pneumocystis jiroveci en pacientes con VIH. Neumol Cir Torax. 2011;70:165 -71.
2. Kanne JP, Yandow DR, Meyer CA. Pneumocystis jiroveci pneumonia: high-resolution CT findings in patients with and without HIV infection. AJR Am J Roentgenol. 2012;198:W555 -61.
3. Lichtenberger JP 3rd, Sharma A, Zachary KC, Krishnam MS, Greene RE, Shepard JA, et al. What a differential a virus makes: a practical approach to thoracic imaging findings in the context of HIV infection: part 1, pulmonary findings. AJR Am J Roentgenol. 2012;198:1295 -304.
Agujeros parietales gigantes
Foramina parietalia permagna
Estimada Editora:
Los agujeros parietales son pequeñas formaciones ubicadas en la calota craneana sobre los huesos parietales, en topografía parasagital. Estos orificios miden en promedio 1 mm, por lo que si superan los 5 mm, se denominan agujeros parietales gigantes (APG). Se asocian a distintos tipos de craneosinostosis, displasias corticales, microcefalia, defectos oculares o del oído, sindactilia y polidactilia con hipoplasia distal de la clavícula y ausencia del acromion, retraso mental, exostosis múltiples, disostosis craneofacial y alteraciones de los vasos intracraneales1,2. Muy rara vez se ha citado su vínculo con malformaciones cardíacas3.
Presentamos el caso de una niña de 3 meses de vida, que había nacido pretérmino (31 semanas) con un peso 1470 gramos. Al nacer, había requerido reanimación cardio-pulmonar y tuvo una internación de 63 días en Neonatología por una comunicación interventricular (CIV) perimembranosa moderada y una comunicación interauricular (CIA) tipo ostium secundum. Fue derivada a nuestra institución para el tratamiento de su malformación cardíaca.
Al ingreso la paciente se encontraba sin fiebre, irritable y con distrés respiratorio. Las fontanelas anterior y posterior eran normales y no presentaban tensión. Sin embargo, detrás de la fontanela anterior se observó una región de partes blandas sobreelevada y de morfología ovalada, que a la palpación se revelaba renitente y con un borde óseo que la delimitaba. Dados los antecedentes cardíacos y los hallazgos en la calota craneana, se solicitó una ecografía transfontanelar (ETF) y una tomografía computada (TC) para descartar alteraciones sindrómicas. En nuestra institución no se llevó a cabo radiografía de cráneo.
La ETF evidenció un extenso defecto óseo con una leve protrusión meníngea y del espacio aracnoideo a través de la diastasis ósea parietal, sin otras alteraciones encefálicas (fig. 1). Por su parte, la TC advirtió mejor el extenso defecto en la conformación de ambos huesos parietales (fig. 2), con una gran solución de continuidad y una amplia zona sin revestimiento óseo a nivel de la calota craneana, que creaba una ‘‘pseudofontanela posterior'' (fig. 3) por donde protruían el espacio aracnoideo y las meninges, un hallazgo compatible con APG. No se observaron otras alteraciones de las estructuras extra e intracraneales, pero en la radiografía de tórax se apreció la típica imagen de corazón en bota de las cardiopatías (fig. 4). La paciente fue tratada por su patología cardíaca y dada de alta para control en su localidad.

Figura 1 Ecografía transfontanelar a través del agujero parietal gigante en la que se observan las meninges y el espacio aracnoideo de características normales.

Figura 2 Tomografía computada de cráneo en ventana parénquima con reconstrucción coronal a través del agujero parietal gigante (cabezas de flecha) donde se evidencia una leve protrusión del espacio aracnoideo y de las meninges.

Figura 3 Tomografía computada de cráneo, en reconstrucción tridimensional, en vista superior, en la que se observa el agujero magno (asterisco) a través de la fontanela anterior (flechas) y, por detrás de ella, una deformidad de ambos huesos parietales, creando una pseudofontanela llamada agujero parietal gigante (cabezas de flecha).

Figura 4 Radiografía de tórax portátil en la que se advierte un aumento de la aurícula (flecha) y el ventrículo derechos (cabeza de flecha), configurando el corazón en bota.
El hueso parietal presenta, en porcentaje variado, pequeños orificios que pueden ser únicos, en par (uno a cada lado), triples (dos de un lado y uno del otro) o pares a cada lado de la sutura sagital, cercanos al Lambda y conocidos como forámenes o agujeros parietales4. Estos permiten el pasaje de venas emisarias que comunican con el seno longitudinal superior. La frecuencia del foramen parietal bilateral alcanza el 59% y el diámetro promedio oscila entre los 0,6 y 1,5 mm 5. Si su diámetro excede los 5 mm, se llaman agujeros parietales gigantes o foramina parietalia permagna. Estos no constituyen simplemente un agujero parietal aumentado de tamaño, sino una patología benigna de defecto en la osificación6.
El tratamiento quirúrgico puede ser necesario cuando el diámetro es muy grande o persiste aumentado de tamaño con el crecimiento del niño. En ambos casos se busca, por medio de injertos óseos autólogos o mallas, proteger el cerebro subyacente7. Los agujeros suelen tener formas variadas, pudiendo ser ovales, redondos, rectangulares, irregulares o con depresiones. La multiplicidad de tamaños, la ausencia de manifestaciones clínicas y la falta de asociación sindrómica conduce a un diagnóstico tardío.
La entidad también es conocida como Catlin mark. Su nombre se debe al apellido de la familia en la que Goldsmith8 describió por primera vez la presencia de esta alteración en el cráneo de varios integrantes del mismo grupo familiar, demostrando así su relación hereditaria. Estudios posteriores han confirmado su herencia autosómica dominante debida a mutaciones heterocigotas en el gen MSX29,10.
Los APG constituyen una condición benigna que puede estar asociada a múltiples alteraciones morfológicas o genéticas. Conocer su clínica y características radiológicas resulta relevante, ya que ante este hallazgo hay que estudiar ampliamente al paciente y su familia.
Atentamente, los autores
Confidencialidad de los datos
Los autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
D.A. Benítez* y A. Surur
Servicio de Diagnóstico por Imágenes, Sanatorio Allende, Córdoba, Argentina
*Autor para correspondencia.
Correo electrónico: dntsc@hotmail.com (D.A. Benítez).
1. Oliva Rodríguez-Pastor S, Camacho Alonso JM, González Gómez JM, Cano España J, Calvo Macías C. Agujeros parietales. An Esp Pediatr. 2001;55:387 -8.
2. Reddy AT, Hedlund GL, Percy AK. Enlarged parietal foramina: association with cerebral venous and cortical anomalies. Neurology. 2000;54:1175 -8.
3. Blanco del Val A, Carrascal Arranz MI, Centeno Malfaz F, Marcos Andrés H, Alcalde Martín C. Foramina parietalia permagna, un raro defecto congénito. An Pediatr. 2003;58:506 -7.
4. Testut L, Latarjet A. Tratado de anatomía humana. 9a edición Barcelona: Salvat; 1971. p. 119 -306.
5. Collipal E, Silva H, Quintas F, Martínez C, Del Sol M. Estudio morfométrico del foramen parietal. Int J Morphol. 2009;27: 481 -4.
6. Tubbs RS, Smyth MD, Oakes WJ. Parietal foramina are not synonymous with giant parietal foramina. Pediatr Neurosurg. 2003;39:216 -7.
7. Kortesis B, Richards T, David L, Glazier S, Argenta L. Surgical managementof foramina parietalia permagna. J Craniofac Surg. 2003;14:538 -44.
8. Goldsmith WM. The Catlin mark: the inheritance of an unusual opening in the parietal bones. J Hered. 1922;13:69 -71.
9. Fein JM, Brinker RA. Evolution and significance of giant parietal foramina. Report of five cases in one family. J Neurosurg. 1972;37:487 -92.
10. Wu YQ, Badano JL, McCaskill C, Vogel H, Potocki L, Shaffer LG. Haploinsufficiency of ALX4 as a potential cause of parietal foramina in the 11p11. 2 contiguous gene deletion syndrome. Am J Hum Genet. 2000;67:1327 -32.

