










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista argentina de radiología
versión On-line ISSN 1852-9992
Rev. argent. radiol. vol.82 no.3 Ciudad Autónoma de Buenos Aires set. 2018
http://dx.doi.org/https://doi.org/10.1055/s-0038-1656560
DOI https://doi.org/10.1055/s-0038-1656560
Pictorial Essay | Ensayo Iconográfico
SÃndromes de neoplasia endocrina múltiple: revisión radiológica
Multiple Endocrine Neoplasia Syndromes: Radiological Review
Ignacio MartÃn GarcÃa1 Rosario Vidal Tocino2 Elisabeth MartÃn GarcÃa3 Manuel Ãngel MartÃn Pérez1 José MartÃn MarÃn BalbÃn1
1 Servicio de Radiodiagnóstico, Complejo Asistencial de Zamora, Zamora, España
2 Unidad de Consejo Genético, Servicio de OncologÃa Médica, Hospital Universitario de Salamanca, Salamanca, España
3 Servicio de OftalmologÃa, Complejo Asistencial de Zamora, Zamora, España
Address for correspondence
Ignacio MartÃn GarcÃa, PhD, Servicio de Radiodiagnóstico, Complejo Asistencial de Zamora, Complejo Asistencial de Zamora, Av. de Requejo 35, 49022 Zamora, España (e-mail: nachetemaga@hotmail.com).
Received:
November 29, 2017
accepted:
April 8, 2018
published online:
June 1, 2018
DOI https://doi.org/10.1055/s-0038-1656560
ISSN 1853-5844
Copyright © 2018, Sociedad Argentina de RadiologÃa. Publicado por Thieme Revinter Publicações Ltda., Rio de Janeiro, Brazil. Todos los derechos reservados.
Resumen
Los sÃndromes de neoplasia endocrina múltiple (MEN), incluyen una serie de enfermedades con alteraciones genéticas que se caracterizan por la presencia de tumores que afectan a dos o más glándulas endocrinas. Son sÃndromes con una herencia autosómica dominante e incluyen tres patrones: MEN 1 (sÃndrome de Wermer), MEN 2 (que incluye MEN 2A o sÃndrome de Sipple y MEN 2B o sÃndrome de Wagenmann-Froboese) y MEN 4. Los adenomas paratiroideos y el carcinoma medular tiroideo, son los tumores más frecuentes del MEN tipo 1 y 2 respectivamente. Esos sÃndromes son más comunes en pacientes jóvenes, con patologÃa de afectación bilateral, múltiple o multifocal y, sobre todo, en pacientes con antecedentes familiares. Es necesario el trabajo en equipo de endocrinólogos, cirujanos, oncólogos y radiólogos para optimizar el tratamiento de esos pacientes.
Palabras Clave: Neoplasia Endocrina Múltiple; Men 1; Men 2; Carcinoma Medular De Tiroides; Feocromocitoma
Abstract
Multiple endocrine neoplasia (MEN) encompasses a serial of familial genetically disorders in wich tumors simultaneusly occur in two or more endocrine organs. MEN sÃndromes are autosomal-dominant disorders categorized into three main patterns: MEN 1 (Wermer syndrome), MEN 2 (includes MEN 2A o Sipple syndrome and MEN 2B o Wagenmann-Froboese syndrome) and MEN 4. Parathyroid adenomas and medullary thyroid carcinoma are the most frecuent tumors in MEN 1 and MEN 2 respectively. These entities will be suspected in younger patients, bilateral, multiple or multifocal disease and, specially, in patients with family background. Cooperation between endocrinologist, surgeons, oncologists and radiologists is pivotal for optimizing patient treatment.
Keywords: Multiple Endocrine Neoplasia; Men 1; Men 2; Medullary Thyroid Carcinoma; Pheochromocytoma
Introducción
Los sÃndromes de neoplasia endocrina múltiple (MEN), incluyen una serie de enfermedades con alteraciones genéticas que se caracterizan por la presencia de tumores que afectan a 2 o más glándulas endocrinas. Son sÃndromes con un herencia autosómica dominante e incluyen tres patrones: MEN 1, MEN 2 y MEN 4. Cada uno de los tipos, se caracteriza por la presencia de tumores en órganos especÃficos. En este artÃculo, describiremos las caracterÃsticas clÃnicas de cada uno de los tipos y mostraremos sus principales hallazgos por imagen.
MEN 1
El MEN tipo 1 es una afección hereditaria autosómica dominante infrecuente (incidencia de 2 casos cada 100.000). La base genética del MEN 1 es una mutación de pérdida de función de la lÃnea germinal del gen MEN 1, que se encuentra en la banda cromosómica 11q13.1 Combina la aparición de tumores en la glándula paratiroides, páncreas y adenohipófisis. El diagnóstico es suficiente con la presencia de 2 de las 3 neoplasias principales. Algunos pacientes desarrollan tumores corticosuprarrenales, carcinoides, angiofibromas faciales, colagenomas y lipomas (â–ºFig. 1).2,3,4
Fig. 1 Espectro de afectación en el sÃndrome de neoplasia endocrina múltiple (MEN) tipo 1.
Tumores Paratiroideos
Los tumores paratiroideos son la primera manifestación de ese sÃndrome en más del 85% de los afectados. La afectación multiglandular no siempre es sincrónica.5,6 En los pacientes con hipercalcemia persistente tras la cirugÃa, la imagen tiene un papel muy importante.7 El tratamiento es quirúrgico desde la paratiroidectomÃa subtotal a la total con reimplante de una glándula en el bazo.8
Hallazgos Radiológicos
El estudio inicial ante sospecha de adenoma paratiroideo se realiza con ecografÃa. Los adenomas paratiroideos se visualizan como lesiones ovaladas, bien delimitadas e hipoecoicas localizados detrás de la glándula tiroides y con aporte arterial en el polo superior o inferior de la glándula paratiroides, generalmente tributario de la arteria tiroidea inferior. La ecografÃa permite, además, ser guÃa en procedimientos intervencionistas como la punción aspiración con aguja fina (PAAF), que contribuye al diagnóstico anatomopatológico de la lesión.
La tomografÃa computada (TC), permite localizar glándulas ectópicas en el mediastino y detrás de la tráquea. La resonancia magnética (RM) posee mayor sensibilidad que la TC para la localización de los adenomas pero no se suele utilizar en primera lÃnea. Los adenomas se visualizan hiperintensos en secuencias potenciadas en T2 e hipointensos en secuencias potenciadas en T1.
En medicina nuclear, el empleo de gammagrafÃa con sestamibi obteniendo imágenes tempranas (15 minutos) y tardÃas (2 horas) tras la inyección del radiotrazador tienen una alta sensibilidad para la detección deadenomas(â–ºFig. 2).5,7

Fig. 2 Adenoma de paratiroides en un paciente con MEN tipo 2. En la ecografÃa imagen transversal (A) y longitudinal (B) se visualizó una lesión redondeada, de contornos bien delimitados e hipoecoica con aporte arterial en su polo superior (C). En la tomografÃa computada (TC) con contraste intravenoso, corte coronal (D) se objetivó una lesión nodular hipercaptante en localización posterior a la glándula tiroidea.
La gammagrafÃa con sestamibi, fase precoz (E) muestra una lesión que captó trazador en la misma localización que la imagen b, compatible con adenoma paratiroideo.
Tumores Neuroendocrinos Pancreáticos
Aparecen en el 80% de los pacientes. El gastrinoma es el tumor neuroendocrino más frecuente en el MEN y es el principal condicionante pronóstico por su potencial maligno. Otros menos frecuentes son los insulinomas y glucagonomas. El principal objetivo de la cirugÃa es evitar o retrasar la aparición de metástasis. La presentación de los gastrinomas suele ser múltiple y de pequeño tamaño, lo que dificulta la resección completa, por eso el conocimiento de la localización duodenal lleva a recomendar la duodenotomÃa, con exploración duodenal sistemática además de la pancreática.6,8
Hallazgos Radiológicos
La ecografÃa no suele ser efectiva porque no permite su visualización y se emplea durante la cirugÃa tras la palpación. Las lesiones se aprecian homogéneas e hipoecoicas. La TC suele ser la técnica de elección tanto para la extensión local como a distancia. Suelen ser lesiones de pequeño tamaño y múltiples por lo que se requiere el empleo de agua como contraste oral. Sin contraste intravenoso, las lesiones son isodensas, con intenso realce en fase arterial por lo que es necesario realizar una fase arterial a los 25-30 segundos tras la inyección de contraste ya que en fase pancreática a los 35-40 segundos o en fase portal de 60-70 segundos pueden ser isodensos con el parénquima pancreático y no ser visibles (â–ºFig. 3). Las metástasis hepáticas suelen ser hipervasculares en fase arterial.

Fig. 3 Insulinoma en un paciente con MEN tipo 1. La TC abdominal sin contraste intravenoso (A) mostró una lesión redondeada isodensa con el parénquima pancreático que deforma su contorno (flecha). En la TC abdominal con contraste, fase arterial (B) se visualizó la lesión hipercaptante localizada en cabeza pancreática. En la TC abdominal con contraste, fase portal (C) la lesión mostró lavado de contraste, mostrándose levemente hiperdensa respecto el parénquima pancreático.
La RM permite aumentar la sensibilidad para detectar esos tumores, se visualizan hipointensos en secuencias potenciadas en T1 e hiperintensos en secuencias potenciadas en T2 con un patrón de captación de contraste similar a la TC. Por otro lado, la gammagrafÃa de receptores de somatostatina con Indio 111, es una técnica que permite obtener imágenes de cuerpo entero para buscar tumores neuroendocrinos ocultos, desafortunadamente no todos los tumores neuroendocrinos expresan receptores de somatostatina. Esa técnica permite conocer si los pacientes responderán a la terapia de radionucleidos y permite monitorizar la respuesta. Por último, la tomografÃa por emisión de positrones (PET), no tiene un rol importante en esa patologÃa ya que se tratan de tumores muy indiferenciados y con un metabolismo bajo por lo que no se visualiza captación de fluorodesoxiglucosa (FDG).5,7
Tumores Adenohipofisarios
Aparecen en el 30% de los pacientes y suelen ser funcionantes (60% prolactinomas, 25% segregan hormona somatotropa y 5% segregan hormona corticotropa), además de la clÃnica derivada por la secreción excesiva de hormonas, pueden provocar clÃnica derivada por la compresión en tumores de gran tamaño.6,7
Hallazgos Radiológicos
La RM es la prueba de elección para visualizar la hipófisis. Se tienen que emplearcampos de visión pequeños con secuencias potenciadas en T1, en los planos sagital y coronal, asà como un estudio dinámico con contraste. Los microadenomas pueden ser de difÃcil visualización por lo quehay que prestar atención a signos indirectos como la convexidad del margen superior dela glándula y erosión de la silla turca. Esas lesiones suelen verse hipointensas en secuencias potenciadas en T1 pre y poscontraste (â–ºFig. 4).2,5,7
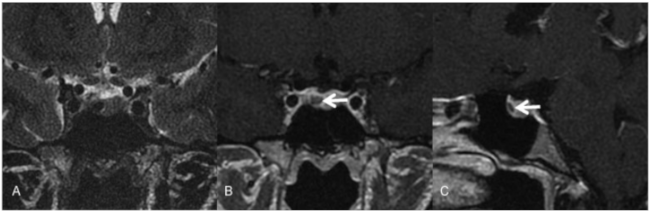
Fig. 4 Adenoma de hipófisis en un paciente con MEN tipo 1. Imágenes de resonancia magnética (RM). En la secuencia potenciada en T2 en el plano coronal se objetivó una lesión redondeada hipointensa en la porción derecha de la adenohipófisis (flecha). Las imágenes con gadolinio en el plano coronal (B) y sagital (C) mostraron la lesión nodular no captante.
Tumores Corticosuprarrenales
Suelen ser adenomassuprarrenales no funcionantes y aparecen hasta en el 40% de los pacientes con MEN 1. Los tumores menores de 4 cm no suelen requerir cirugÃa, en cambio los tumores funcionantes pueden requerir adrenelectomÃa.5
Hallazgos Radiológicos
Aproximadamente un 70% a un 80% de los adenomas, son ricos en lÃpidos intracelulares. En la TC sin contraste, presentan valores densitométricos inferiores a las 10 UH. El estudio dinámico con contraste debe tener una fase portal y otra tardÃa, además del estudio sin contraste. Los adenomas presentan un lavado relativo mayor del 40% y un lavado absoluto mayor al 60%. La secuencia potenciada en T1 fase/ fuera de fase permite identificar las lesiones con lÃpidos intracelulares (â–ºFig. 5).
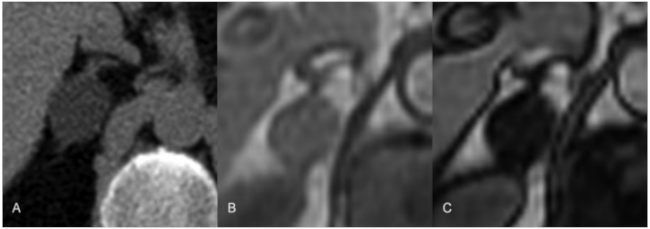
Fig. 5 Adenoma suprarrenal en un paciente con MEN tipo 1. La TC sin contraste intravenoso (A) mostró una lesión nodular en glándula suprarrenal derecha con valores densitométricos inferiores a 10UH. En las imágenes de resonancia magnética (RM) en fase (B) y fase opuesta se objetivó pérdida de intensidad de la señal en fase opuesta, caracterÃstica de los adenomas suprarrenales.
Los adenomas pobres en lÃpidos intracelulares son difÃciles de caracterizar por presentar valores superiores a las 10 UH o por falta de desplazamiento quÃmico en la secuencia de fase opuesta. Por otro lado, los carcinomas suprarrenales son lesiones muy raras y presentan caracterÃsticas de agresividad con necrosis, hemorragia y tendencia a invadir estructuras vecinas.5,7
MEN 2
El MEN 2 es una afección hereditaria autosómica dominante con una prevalencia estimada de 1 por cada 30000 personas, afectando por igual a ambos sexos. La base genética del MEN 2 se debe a una mutación en la lÃnea germinal del protooncogen RET situado en el cromosoma 10q11.2.9 Se caracteriza por la aparición de carcinoma medular de tiroides, feocromocitoma e hiperparatiroidismo. Se divide en tres subtipos (â–ºFig. 6):

Fig. 6 Espectro de afectación en el sÃndrome MEN tipo 2.
MEN 2A: en prácticamente todos los afectados existe carcinoma medular de tiroides, feocromocitomas aproximadamente en el 50% de los afectados e hiperparatiroidismo en el 10% al 30% de los afectados. La enfermedad de Hirschsprung y el liquen cutáneo amiloidótico, se pueden asociar en esos pacientes.
MEN 2B: similar al MEN 2A pero sin hiperparatiroidismo, presentan un hábito marfanoide y además pueden desarrollar ganglioneuromas en el tracto intestinal y neuromas en otras localizaciones como la lengua (►Fig. 7).2,10 Las manifestaciones oftalmológicas descritas más frecuentemente en los pacientes son el engrosamiento de los nervios corneales, cejas prominentes asociadas a neuromas en los párpados, neuromas subconjuntivales, dificultad para la dilatación pupilar y elevación de la presión intraocular.11
Carcinoma medular tiroideo familiar: Sin desarrollo de feocromocitoma ni hiperparatiroidismo primario.

Fig. 7 FotografÃas de un paciente con MEN tipo 2B. (A) Neuroma perilÃmbico (flecha). (B) Neuromas linguales (flecha).
Carcinoma Medular de Tiroides
Es el primer tumor que se desarrolla en pacientes con MEN 2 y es la causa más común de fallecimiento en esos pacientes.9 El carcinoma medular se origina en las células parafoliculares o células C. La afectación suele ser bilateral y multifocal además de tener un peor pronóstico y aparecer tempranamente.10
La clÃnica puede debutar con la palpación de un nódulo tiroideo o adenopatÃa y en algunos casos se llega al diagnóstico por la clÃnica del feocromocitoma o del hiperparatiroidismo concomitante.12 El diagnóstico bioquÃmico se realiza con la determinación de calcitonina, siendo un excelente marcador de la enfermedad que permite predecir la respuesta al tratamiento. El antÃgeno carcinomenbrionario (CEA), puede ser útil en los pacientes con carcinoma metastásico poco diferenciado que no eleva las cifras de calcitonina.12,13
Hallazgos Radiológicos
La ecografÃa tiroidea es la prueba de elección en la valoración inicial del carcinoma medular. Las lesiones presentan caracterÃsticas de malignidad: hipoecogenicidad, mala delimitación de sus márgenes, vascularización interna y micro o macrocalcificaciones. Las calcificaciones son los hallazgos más significativos de malignidad en el carcinoma medular. Además, hay que tener en cuenta que el diámetro longitudinal mayor que el transversal es predictivo de malignidad. La ecografÃa además permite detectar adenomas en pacientes con sÃndrome MEN 2A.
La invasión local es común, con una diseminación ganglionar en el 50% de los casos. La afectación a distancia es, por orden de frecuencia, en el hÃgado (49%-62%), hueso (40%-74%) y pulmón (33%-35%), ocurre entre el 15% y el 25% de los casos. Por ello se recomienda la realización preoperatoria de TC de cuello, tórax y abdomen para el estadiaje en los pacientes con adenopatÃas. Los hallazgos en la TC no son especÃficos en el caso del tumor primario y en las adenopatÃas veremos calcificaciones densas e irregulares (â–ºFig. 8). La TC de abdomen permitirá visualizar la existencia de feocromocitoma, que deberá ser extirpado previamente a cualquier otra cirugÃa.

Fig. 8 Carcinoma medular de tiroides en paciente con MEN tipo 2 con adenoma paratiroideo asociado (asterisco). La ecografÃa, corte axial (A) mostró una lesión hipoecoica, con márgenes mal delimitados y con microcalcificaciones (flecha), el corte longitudinal (B) con doppler color mostró una lesión sin flujo significativo (flecha). En la TC torácica con contraste intravenoso se objetivaron adenopatÃas a nivel paratraqueal alto (B) y paratraqueal bajo (C) con múltiples calcificaciones densas e irregulares caracterÃsticas de la diseminación ganglionar en esa enfermedad.
Las metástasis hepáticas pueden ser hiper o hipoecogénicas en la ecografÃa. Suelen ser lesiones hipervasculares, con degeneración quÃstica y en algunos casos de pequeño tamaño y pueden calcificar. Las lesiones óseas pueden ser lÃticas, blásticas o mixtas. Las metástasis pulmonares pueden ser micro o macronodulares, se ha descrito afectación miliar bilateral en algunos casos y lesiones perihiliares o conglomerados calcificados.
La sensibilidad de la PET/TC es variable dependiendo del grado de diferenciación del tumor siendo menos sensible en tumores muy indiferenciados y en pacientes con lesiones metastásicas de pequeño tamaño, en cambio es más sensible en pacientes con niveles altos de calcitonina (> 1000 pg/mL). El empleo de nuevos trazadores, como la 18F-dihidroxifenilalanina, permiten una mayor sensibilidad que la FDG. La PET/TC puede tener un papel importante en los pacientes con carcinoma recurrente.12,14
Feocromocitoma
El feocromocitoma es un tumor que se origina en los paraganglios en la médula adrenal y suele ser bilateral. Pueden ser asintomáticos o tener clÃnica relacionada con hipertensión arterial. El diagnóstico inicial es bioquÃmico con la medición en orina de adrenalina, noradrenalina, metanefrinas o normetanefrinas. Posteriormente, se emplean pruebas de imagen para su localización.1,10
Hallazgos Radiológicos
La ecografÃa es la técnica inicial ante la sospecha de feocromocitoma y permite valorar alteraciones morfológicas y de tamaño de las glándulas suprarrenales, aunque debido a limitaciones dependientes del operador, a causa del propio paciente, como la obesidad, o al tratarse de tumores de pequeño tamaño, puede tener una menor sensibilidad que la TC o RM. Se puede visualizar como una lesión de contornos bien delimitados, ovoidea y de ecogenicidad heterogénea.
En la TC, los feocromocitomas pueden mostrar calcificaciones en el estudio sin contraste y mostrar un intenso realce tras el contraste, también se pueden observar áreas de necrosis en algunas lesiones. El empleo de contrastes yodados iónicos puede desencadenar una crisis hipertensiva, por eso están contraindicados. Los contrastes no iónicos no conllevan el mismo riesgo y pueden ser empleados sin bloqueo alfa adrenérgico.
La RM es una prueba muy sensible para la detección de feocromocitoma siendo la prueba de elección para su detección debido a la ausencia de radiación ionizante. Esas lesiones se visualizan hipointensas en la secuencia potenciada en T1 y muy hiperintensas en la secuencia potenciada en T2 con zonas heterogéneas por la existencia de necrosis, hemorragia o calcificación. Tras la inyección de gadolinio realzan de manera intensa (►Fig. 9).
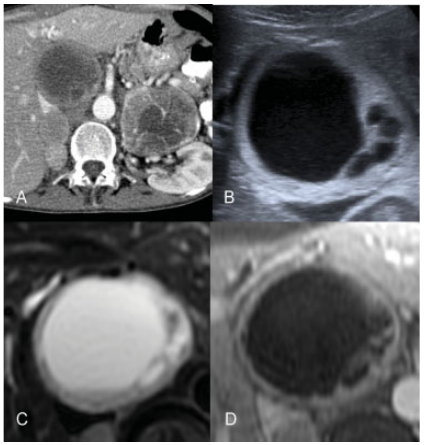
Fig. 9 Feocromocitomas en paciente con MEN tipo 2. En la TC con contraste intravenoso (A) se identificaron feocromocitomas bilaterales. En la ecografÃa (B) se apreció feocromocitoma derecho ampliado con zonas quÃsticas y capsula gruesa. En la RM se identificó la misma lesión que en ecografÃa, lesión encapsulada y heterogénea que depende de glándula suprarrenal derecha que en la secuencia potenciada en T2 (C) presentó un aspecto heterogéneo objetivándose marcadamente hiperintensa, y con captación de predominio periférico en la secuencia con gadolinio (D).
La gammagrafÃa con metayodobencilguanidina (MIBG) de cuerpo entero, es más sensible que la TC y la RM para la detección de hiperplasia medular adrenal, la cual se cree que es la precursora del feocromocitoma y podrÃa ser tratada con adrenelectomÃa en pacientes con MEN 2. El empleo de nuevos trazadores con afinidad por el sistema nervioso simpático, como la 11C-hidroxiefedrina o la 18F-dihidroxifenilalanina, aumenta la sensibilidad para la detección del feocromocitoma.5,7
Hiperparatiroidismo Primario
Aparece aproximadamente entre el 10% y el 30% de los pacientes MEN 2, pero es muy raro que sea la manifestación inicial. La hiperplasia paratiroidea no se diferencia de la hiperplasia de los casos esporádicos. Generalmente hay un aumento de tamaño de las cuatro glándulas.10
MEN 4
El MEN 4 es una forma recientemente descrita del espectro MEN con tumores paratiroideos y de la adenohipófisis, también pueden desarrollar carcinoides gástricos y bronquiales o gastrinomas.5
Confidencialidad de los Datos
Los autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio, han recibido información suficiente y han dado su consentimiento informado por escrito.
Conflicto de Intereses
Los autores declaran no tener ningún conflicto de interés.
Contribución de AutorÃa
Todos los firmantes en la carta de presentación han intervenido en la concepción del estudio, en el diseño, en la obtención de datos, análisis e interpretación. También han colaborado en la redacción y revisión crÃtica. Todos los autores aprueban la versión remitida del manuscrito.
BibliografÃa
1 Quesada Charneco M. Hiperparatiroidismo primario en situaciones especiales: sÃndromes de neoplasia endocrina múltiple y cáncer de paratiroides. Endocrinol Nutr 2009;56 (Suppl 1):35-40 [ Links ]
2 Shinagare AB, Giardino AA, Jagannathan JP, Van den Abbeele AD, Ramaiya NH. Hereditary cáncer sÃndromes: a radiologist’sperspective. AJR Am J Roentgenol 2011;197:1001-1007
3 Sato M, Kihara M, Nishitani A, et al. Large and asymptomatic pancreatic islet cell tumor in a patient with multiple endocrine neoplasia type 1. Endocrine 2000;13(03):263-266 [ Links ]
4 Thakker RV. Multiple endocrine neoplasia type 1 (MEN1). Best Pract Res Clin Endocrinol Metab 2010;24(03):355-370 [ Links ]
5 Grajo JR, Paspulati RM, Sahani DV, Kambadakone A. Multiple Endocrine Neoplasia Syndromes: A Comprehensive Imaging Review. Radiol Clin North Am 2016;54(03):441-451 [ Links ]
6 Thakker RV, Newey PJ, Walls GV, et al; Endocrine Society. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 2012;97(09):2990-3011 [ Links ]
7 Scarsbrook AF, Thakker RV, Wass JAH, Gleeson FV, Phillips RR. Multiple endocrine neoplasia: spectrum of radiologic appearances and discussion of a multitechnique imaging approach. Radiographics 2006;26(02):433-451 [ Links ]
8 Familiar C, Antón T, Moraga I, Ramos A, Marco A. A propósito de un caso de neoplasia endocrina múltiple tipo 1. Revisión de algunas manifestaciones clÃnicas y controversias en el tratamiento. Endocrinol Nutr 2011;58(02):84-89 [ Links ]
9 Ohyama T, Sato M, Murao K, et al. A case of multiple endocrine neoplasia type 2B undiagnosed for many years despite its typical phenotype. Endocrine 2001;15(02):143-146 [ Links ]
10 Wohllk N, Schweizer H, Erlic Z, et al. Multiple endocrine neoplasia type 2. Best Pract Res Clin Endocrinol Metab 2010;24(03): 371-387 [ Links ]
11 Puvanachandra N, Aroichane M. Diffuse corneoscleral limbal neuromas with prominent corneal nerves in multiple endocrine neoplasia syndrome type IIB. J Pediatr Ophthalmol Strabismus 2010;47(03):171-173 [ Links ]
12 Ganeshan D, Paulson E, Duran C, Cabanillas ME, Busaidy NL, Charnsangavej C. Current update on medullary thyroid carcinoma. AJR Am J Roentgenol 2013;201(06):W867-W876 [ Links ]
13 Takami H. Medullary thyroid carcinoma and multiple endocrine neoplasia type 2. Endocr Pathol 2003;14(02):123-131 [ Links ]
14 Patel CN, Chowdhury FU, Scarsbrook AF. Clinical utility of hybrid SPECT-CT in endocrine neoplasia. AJR Am J Roentgenol 2008;190 (03):815-824 [ Links ]

