











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkEstimados editores:
La encefalitis de Rasmussen (ER) es un tipo de encefalopatía epiléptica descripta por el neurocirujano Theodore Rasmussen (Fig. 1) y colegas en 1950.1,2,3

Es un trastorno neurológico raro subagudo/crónico, de origen desconocido (posiblemente secundario a daño neuronal inmunomediado), asociado a epilepsia focal que determina la unilateralidad.1,3,4,5,6,7,8,9,10
Su prevalencia es de 0-18 por 100.000 personas. Su distribución según sexo, predominio geográfico y étnico no han sido reportados. Inicia entre los 3 y 6 años de edad (90%), en un niño previamente normal, con convulsiones focales progresivas -característicamente con epilepsia parcialis continua (EPC)-, y posterior deterioro neurológico paulatino y hemiparesia. La epilepsia que ocasiona es resistente a fármacos.1,3,5
El presente artículo tiene por objetivo revisar y destacar el aporte de las neuroimágenes en el diagnóstico preciso y oportuno de la ER, que facilite un abordaje precoz de esa patología.
Se presenta el caso de una paciente de 9 años de edad, con historia perinatológica normal. Presentó epilepsia refractaria al tratamiento farmacológico desde los 3 años. Debido a la ausencia de respuesta a fármacos, se indicó tratamiento quirúrgico de hemisferectomía derecha (desconexión hemisférica funcional) a los 4 años de edad. Actualmente, se encuentra en internación domiciliaria y en estado de postración crónica, con empeoramiento en los últimos cinco días, asociado a hemiplejía facio-braquio-crural izquierda e incontinencia urinaria.
En la resonancia magnética (RM) encefálica se identifican cambios anatómicos postquirúrgicos a nivel de la convexidad derecha (Fig. 2A). Subyacente a esa zona, se observa pérdida de volumen del hemisferio cerebral (Fig. 2B), con áreas de afinamiento cortical (Fig. 3A), asociadas a signos de gliosis evidentes en secuencias ponderadas en T2 y FLAIR, y aumento de volumen y deformidad del sistema ventricular supratentorial, por leve tracción (Fig. 3A). En todas las secuencias, se visualiza área de encefalomalacia multiquística córtico-subcortical fronto-témporo-parietal derecha (Fig. 3A), en cuyo interior se distingue una pequeña banda de aspecto gliótico secuelar, con cierto refuerzo postcontraste (Fig. 4A) que podría vincularse con convulsión reciente. Se observa, además, pérdida de volumen y cambios de señal del hemisferio cerebeloso izquierdo (diasquisis cruzada; Fig. 3B). En la secuencia difusión (DWI y mapa de ADC) no se identifican focos de restricción (Fig. 4B).
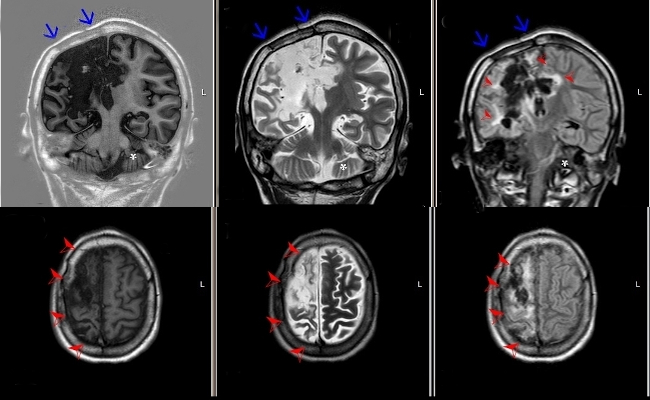
Fig. 2 RM encefálica. Arriba (corte coronal): T1 (izquierda), T2 (centro) y FLAIR (derecha): Signos de instrumentación quirúrgica (flecha azul) y signos de gliosis evidentes en secuencias ponderadas en T2 y FLAIR (puntas de flechas). Abajo (corte axial): T1 (izquierda), T2 (centro) y FLAIR (derecha): Hemiatrofia cerebral derecha (puntas de flechas).
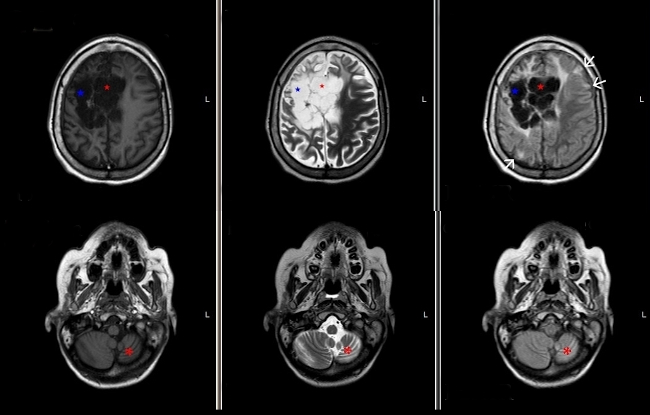
Fig. 3 RM encefálica. Arriba (corte axial): T1 (izquierda), T2 (centro) y FLAIR (derecha) que evidencia encefalomalacia multiquística (asterisco azul) y dismorfismo y dilatación del sistema ventricular supratentorial (asterisco rojo). En secuencias ponderadas en T2/FLAIR se reconocen áreas de afinamiento cortical y lesiones corticales hiperintensas (flecha). Abajo (corte axial): secuencias ponderadas en T1 (izquierda), T2 (centro) y FLAIR (derecha) hemiatrofia cerebelosa izquierda (diasquisis cerebelosa cruzada).
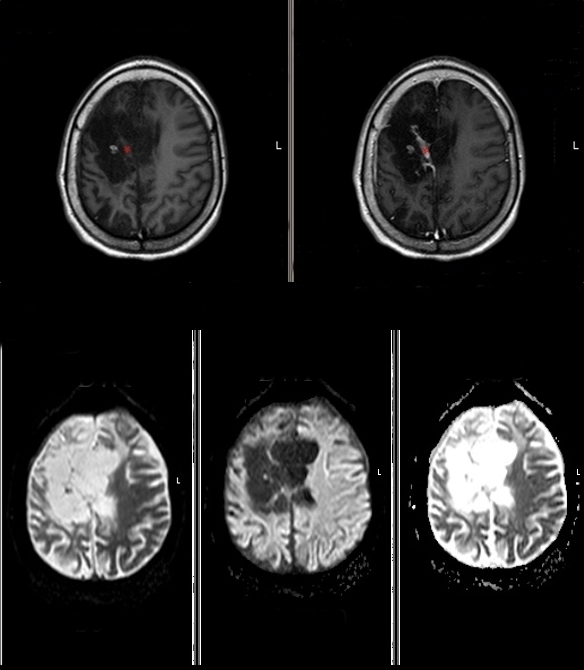
Fig. 4 RM encefálica. Arriba (corte axial): secuencias ponderadas en T1 sin (izquierda) y con gadolínio (derecha) que evidencia banda de gliosis que realza (asterisco). Abajo (corte axial): RMI en difusión en valores b0 (izquierda) y b100 (centro) y mapa ADC (derecha): muestra encefalomalacia multiquística con difusión facilitada con baja señal en DWI y alta señal en ADC.
La ER presenta etapas histológicas similares a la encefalitis viral, que comprenden desde inflamación temprana (Estadio 1) con linfocitos T, gliosis y neuronas sanas, hasta etapas tardías con amplia muerte neuronal, cavitación y vacuolización de la corteza cerebral (Estadio 4).1,3,4,5,6,10
En la ER puede afectarse cualquier área del cerebro, aunque el compromiso es principalmente cortical, con inicio focal y predilección frontocentral o insular, y posterior propagación multifocal unihemisférica ("marcha a través del hemisferio"), con heterogeneidad de etapas histológicas entre los diferentes focos.1,3,4,6,7,9,10
En el año 2005, un consenso europeo propuso criterios formales para el diagnóstico de ER, procurando su detección precoz y limitando la biopsia cerebral a casos puntuales. Deben cumplirse los tres Criterios A, o bien al menos dos Criterios B.3,7
CRITERIOS A:
Criterio clínico
Criterio EEG
RM: Atrofia cortical focal unihemisférica y al menos uno de los siguientes: - Hiperintensidad en sustancia gris o blanca ponderada en T2/FLAIR - Hiperintensidad o atrofia de la cabeza del núcleo caudado ipsilateral
CRITERIOS B:
Criterio clínico
Histopatología compatible
Si no se realiza biopsia: RM con gadolinio: atrofia cortical focal unihemisférica progresiva, sin realce; y tomografía computada (TC) cerebral que excluya calcificaciones debidas a una vasculitis unihemisférica.
A continuación, se resumen los hallazgos posibles de identificar en las neuroimágenes.1,3,4,5,6,7,8,9
TC encefálica: al inicio de las convulsiones puede ser normal; desde el día 7 y durante etapa temprana de ER, inespecífica; desde día 30 y en etapa tardía de ER: atrofia cortical unilateral.5,8
RM: útil para detectar las alteraciones estructurales características, guiar la biopsia cerebral, orientar para eventual tratamiento farmacológico y efectuar el seguimiento de etapa, número y tamaño de lesiones.3,4,9
-T2 y/o FLAIR: más útiles en etapas tempranas (< 1 año): Profundización de cisternas y surcos insulares y peri-insulares; hiperintensidad cortical o subcortical (con o sin edema giral), con inicio focal perisilviano y posterior extensión unihemisférica (aumento del número y/o tamaño de los focos); agrandamiento unilateral del sistema ventricular; atrofia ipsilateral de la cabeza del núcleo caudado y del putamen.1,4,5,6,7,8,9
-T1: más útil en etapas tardías, con evidencia de progresión del cambio de señal y de la hemiatrofia cortical unilateral (a predominio perisilviano); leve atrofia en el hemisferio cerebral no afectado (probablemente por degeneración de fibras comisurales); dilatación ventricular ex-vacuo; diasquisis cerebelosa cruzada.1,3,4,5,6,7,8,9,10
-T1 con contraste: Habitualmente sin realce significativo post-contraste.
DWI/ADC: Puede observarse restricción en áreas con alteración de señal.5
Espectroscopía por RM: disminución de NAA/Cho por pérdida neuronal, y eventual aumento de lactato.7,8
Tomografía de emisión de positrones con fluordeoxiglucosa (PET 18F-FDG): Inicialmente: Área/s hipermetabólica/s focal/es; Posteriormente:
Hipometabolismo cerebral unilateral difuso (excepto en los focos activos): por disminución de actividad neuronal.3,8,9
Entre los diagnósticos diferenciales deben considerarse (además de causas farmacológicas proconvulsivantes, metabólicas, inflamatorio/infecciosas y degenerativas) otros síndromes epilépticos unihemisféricos, entre los que se incluyen: displasia cortical, hemimegalencefalia, esclerosis tuberosa, síndrome de Sturge-Weber, MELAS e infarto hemisférico neonatal (Dyke-Davidoff-Manson).

