











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Las cardiopatías congénitas (CC) abarcan un conjunto de anomalías estructurales y funcionales que se originan durante la embriogénesis y afectan la estructura de las cavidades cardíacas y/o de los grandes vasos. Poseen una prevalencia mundial que oscila entre 6 y 9 por 1000 nacidos vivos1 y constituyen una de las principales causas de muerte prenatal, neonatal e infantil y de morbilidad en edad pediátrica2. La mayoría de las CC se presentan de manera aislada, sin otros defectos asociados, aunque en el 25 al 40% de los casos forman parte de una entidad sindrómica. Los defectos septales, como la comunicación interventricular (CIV) o interauricular (CIA), son las CC más comunes (alrededor del 50%) y constituyen la mayor proporción de los defectos cardíacos aislados3. Las CC del tipo conotroncal (CCC), como la tetralogía de Fallot (TDF), la trasposición de grandes vasos (TGV), la doble salida del ventrículo derecho (DSVD), entre otras, afectan la formación de los tractos de salida y el arco aórtico3, constituyen del 20 al 30% de las CC y son la primera causa de cianosis de origen cardíaco durante el primer año de vida4.
La etiología de las CC es heterogénea. En el 50-70% de los casos no existe una causa identificable para el defecto cardíaco, y se lo considera de etiología multifactorial por la interacción de factores genéticos y ambientales5. Entre las causas ambientales se encuentran los teratógenos, como la exposición materna a alcohol, diabetes pregestacional, talidomida, anticonvulsivantes, agentes infecciosos, etc6.
Entre los factores genéticos, las anomalías cromosómicas se detectan en un 9-18% de los pacientes, y en general en niños que poseen alguna otra anomalía además de la cardíaca7. Algunos ejemplos son las trisomías 21, 18, 13, la monosomía del X y las deleciones 5p o 4p8, entre otras. Las microdeleciones o microduplicaciones de regiones genómicas, comúnmente denominadas CNV (Copy Num-ber Variations), se presentan entre el 5% y el 30% de los pacientes 9-11. Son más frecuentes en pacientes que poseen alguna otra anomalía extracardíaca12, pero también se han identificado en individuos con CC aisladas (CCA)4. La más común es la microdeleción en 22q11 (del22q11), que se observa en 13-18% de los pacientes con CCC13'14.
En el 5-20% de los pacientes con CC se identifican alteraciones en un único gen. Algunas de ellas son causales de síndromes monogénicos, aunque también pueden conducir a la aparición de CCA15. La lista de genes implicados es dinámica, pero se estima que hay alrededor de 450 genes asociados a CC5,16,17. NKX2-5 fue el primer factor de transcripción para el cual se observó una asociación entre variantes génicas y CC 17,18. Es uno de los genes que ha sido más estudiado y en los que se han encontrado más variantes de secuencia en relación con estas patologías19.
Según diversos estudios, la presencia de defectos genéticos se relaciona con el desarrollo de la patología, aunque hasta la fecha no se conocen todas las variantes potencialmente patogénicas. En Argentina, aún son muy limitados los estudios orientados a conocer las diferentes causas genéticas de las CC. Teniendo en cuenta estas observaciones, en el Departamento de Diagnóstico Genético del Centro Nacional de Genética Médica (CNGM) se inició una línea de investigación con el objetivo de analizar regiones genómicas y genes candidatos en un grupo de pacientes argentinos con CC, para detectar variantes genéticas asociadas a la patología. En un estudio multicéntrico preliminar realizado en 2013 en 80 pacientes con CCC, se observó que el 21% poseía deleción 22q11 y el 6%, un desbalance en el brazo corto del cromosoma 1720.
El objetivo del presente trabajo fue evaluar la presencia de anomalías cromosómicas, desbalances genómicos o variantes de secuencias en una muestra de afectados con CC en Argentina y, en un grupo de pacientes con CCC del estudio previo ya referido, abordar específicamente la presencia de variantes en el gen NKX2-5.
MÉTODOS
Se realizó un estudio descriptivo de una serie de casos, en el que se analizaron causas genéticas en pacientes con CC. La población blanco fueron los pacientes de hasta 16 años de edad con CC, atendidos en las instituciones participantes.
Se efectuó un muestreo por conveniencia entre junio de 2015 y febrero de 2019. Se incluyó a pacientes con CC detectados en maternidades de la Región Sanitaria VI de la provincia de Buenos Aires y la maternidad Sardá de la ciudad de Buenos Aires, que participan de la Red Nacional de Anomalías Congénitas (RENAC), así como el Hospital El Cruce Dr. Néstor Carlos Kirchner y el Hospital de Niños Sor María Ludovica, ambos de la provincia de Buenos Aires. Para el estudio específico de variantes en NKX2-5 se incluyó a pacientes con CCC provenientes de cuatro hospitales de Resistencia (Chaco), La Plata (Buenos Aires), Neuquén Capital (Neuquén) y Salta Capital (Salta) en el período comprendido entre mayo de 2013 y mayo de 2014.
Se incluyó a pacientes de hasta 16 años con una anomalía estructural del corazón aislada o asociada a otras anomalías congénitas (AC). Los criterios de exclusión comprendieron: fetos muertos, presencia de ductus en recién nacidos menores a 37 semanas de gestación o de foramen oval y niños con síndrome de Down. Asimismo, quedaron excluidos los pacientes cuyos padres o representantes legales no otorgaron el consentimiento para participar en el estudio.
La muestra incluyó a 289 afectados: 98 con anomalías congénitas múltiples (ACM, 48 niñas y 50 niños) y 191 con CCA (96 niñas y 95 niños). Además, se incluyó a 62 pacientes con CCC (11 con ACM y 51 con CCA) del estudio multicéntrico previo, para los cuales solo se analizaron variantes en el gen NKX2-5.
La variable dependiente del estudio fue la CC con su descripción a campo abierto. Las variables independientes fueron: sexo (femenino, masculino, indeterminado); edad al diagnóstico (expresada en meses postnatales); presencia de otra anomalía congénita asociada (sí/no); anomalía/s congénita/s asociada/s (descripción a campo abierto); presencia de anomalía cromosómica (sí/no); anomalía cromosómica (descripción a campo abierto); presencia de CNV (sí/no); descripción de la CNV (descripción a campo abierto); clasificación de la CNV (ver más abajo); presencia de variantes de secuencia (sí/no); variante de secuencia (descripción a campo abierto); y clasificación de la variante (ver más abajo).
En lo que respecta al algoritmo de estudio, se realizó la anamnesis completa para los afectados incluidos entre 2015 y 2019. A todos los pacientes con ACM se les efectuó el estudio citogenético para determinación del cariotipo, mientras que aquellos que presentaron CCC -ya sea aisladas o con otra anomalía mayor- fueron sometidos a un estudio de amplificación múltiple de sondas dependiente de ligación (MLPA, por sus siglas en inglés) para diagnóstico de deleción 22q11.2. A posteriori, a un subgrupo de pacientes seleccionados con ACM se les realizó estudio de hibridación genómica comparada por microarreglos de ADN (a-CGH, por sus siglas en inglés).
El estudio citogenético se realizó por la técnica de bandeo G (tripsinización y coloración con Wright) en al menos 25 metafases/paciente con un nivel de resolución de 400550 bandas a partir de sangre periférica anticoagulada con heparina y cultivada según protocolos estándar.
La extracción de ADN se llevó a cabo mediante precipitación salina de una muestra de sangre periférica21.
Los desbalances en la región 22q11 y otras 5 regiones genómicas asociadas a CC se evaluaron mediante MLPA utilizando un kit comercial (SALSA MLPA P250, MRC-Holland, Países Bajos) según las instrucciones del fabricante.
Para la evaluación de CNV mediante a-CGH se utilizó un microarreglo comercial (ISCA 8x60K, Agilent) siguiendo las instrucciones del fabricante. Los resultados de la hibridación se analizaron con el programa Cytogenomics (Agilent). Las ganancias y pérdidas detectadas se valoraron como CNV benignas, de significado clínico incierto, probablemente benignas, probablemente patogénicas o patogénicas, teniendo en cuenta las recomendaciones del American College of Medical Genetics and Genomics22.
Para evaluar las variantes de secuencias en el gen NKX2-5, las regiones exónicas del gen y sus regiones intrónicas adyacentes se amplificaron mediante la reacción en cadena de la polimerasa con cebadores (primers) específicos19. Los productos amplificados se secuenciaron por el método de Sanger. La relevancia clínica de las variantes se valoró en forma similar a lo consignado en el apartado anterior por comparación con la secuencia de referencia para el gen23.
El proyecto y su correspondiente consentimiento informado fueron evaluados y aprobados por el Comité de Ética en Investigación de la Administración Nacional de Laboratorios e Institutos de Salud (ANLIS).
RESULTADOS
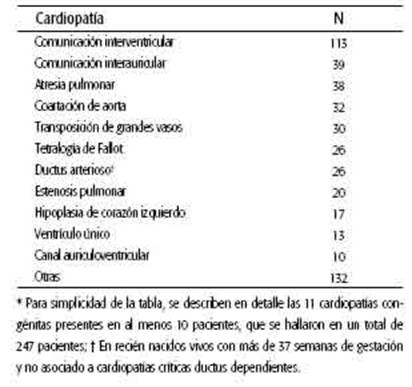
La Tabla 1 resume las cardiopatías que se observaron en la serie de pacientes incluidos entre 2015 y 2019. La más frecuente fue la CIV, presente en 113 pacientes, seguida de CIA y atresia pulmonar (39 y 38 pacientes, respectivamente). En esta serie de pacientes, el número de niños y niñas que ingresaron al estudio fue similar, tanto para aquellos que presentaron ACM como en el caso de CCA. La media de edad fue de 15,9 meses con un error estándar de 2,3 meses, y la mediana de edad al diagnóstico fue de 2,3 meses.
La Tabla 2 detalla aquellos casos que poseían un diagnóstico clínico presuntivo al momento de su inclusión, y la Tabla 3 resume los resultados obtenidos de los estudios citogenéticos realizados en los 98 afectados con ACM. Como se consigna, para 36 pacientes no fue posible realizar en forma completa el estudio. Entre los 62 restantes, 53 poseían un cariotipo normal y 9 presentaron alguna anomalía cromosómica. En el afectado que presentó una translocación aparentemente balanceada entre los cromosomas 1 y 2, se prosiguió con su estudio por a-CGH para estimar si existía alguna pérdida o ganancia submicroscópica de material genético (ver Tabla 4). La paciente que presentó la doble trisomía 47,XXX /47,XX,+ 14 fue detectada por a-CGH ante la dificultad de realizar el primer análisis del cariotipo y confirmada con posterioridad en un segundo análisis citogenético.
En 94/97 pacientes con CCC se completó el análisis de MLPA; 20 pacientes (21%) presentaron la del22q11 de 3 Mb: 7/22 pacientes con ACM (31%) y 13/72 con CCA (18%). Un paciente presentó una deleción intragénica en el exón 7 del gen TBX1.
Con posterioridad, se analizó a 47 pacientes por a-CGH (Tabla 4). En todos los casos, las CNV que fueron clasificadas como patogénicas se corresponden con regiones previamente descriptas en la literatura, y no se halló más de un paciente con una misma CNV. En 5 pacientes se observaron CNV para las cuales aún no se determinó si poseen o no relevancia clínica (VSI: variantes de significado incierto).
El análisis de la secuencia codificante del gen NKX2-5 se presenta en la Tabla 5. Se hallaron 2 variantes noveles: una en un exón que codifica para un cambio sinónimo (no hay cambio de aminoácido) y su posición no está evolutivamente conservada (datos no mostrados); la otra variante novel es intrónica y no modificaría ni generaría sitios conservados de corte y empalme de exones de acuerdo con las predicciones bioinformáticas. Las demás variantes halladas ya fueron descriptas previamente en la población general.


DISCUSIÓN
Las CC constituyen las AC más frecuentes y una de las principales causas de mortalidad perinatal. De acuerdo con los datos de la RENAC, que realiza vigilancia hospitalaria de AC en Argentina24, la prevalencia de nacidos con CC en el período 2009-2018 fue de 4,2/1000 nacimientos. La etiología de las CC es mayoritariamente multifactorial, si bien se describió que existe recurrencia familiar tanto para casos sindrómicos como para casos aislados, consistente con la contribución genética en el desarrollo de la patología5.

TABLA 4: Desbalances hallados en pacientes con anomalías congénitas múltiples estudiados por array-CGH.

En este trabajo se detectaron anomalías cromosómicas en el 11,3% de los pacientes, entre las cuales la trisomía 18 fue la más frecuente. Este porcentaje fue mayor al observado por otros autores si se tiene en cuenta la exclusión de los pacientes con diagnóstico de síndrome de Down25-26. Lamentablemente, en el 36,7% de los pacientes no fue posible determinar su cariotipo por falta de respuesta del cultivo celular. Esta situación es relativamente frecuente en los recién nacidos con ACM, que además tienen muy comprometida su salud (algunos fallecen antes de obtener una nueva muestra).
El síndrome de del22q11, también conocido como velo-cardiofacial/DiGeorge, es el síndrome de microdeleción más común, y las CCC son una de las manifestaciones clínicas más frecuentes. No obstante, se han descripto pacientes con CCC aislada (CCCA) que presentan del22q 1 1 27-28. Por lo tanto, para todos los pacientes de esta cohorte con CCC se analizó la presencia de desbalances en regiones asociadas a CC mediante MLPA. Estos resultados indican que la proporción de pacientes con del22q11 (21%) es similar a la consignada por otros autores29-30, aunque algunos estudios refieren frecuencias más bajas, que oscilan entre 2,5% y 6,8%31-32. Estas diferencias podrían deberse a la proporción de las distintas CCC estudiadas, ya que en pacientes con TDF o tronco arterioso persistente se observa una mayor frecuencia de la deleción que en aquellos con TGV o DSVD33. Asimismo, se ha señalado que pacientes de origen hispánico poseen mayor frecuencia de esta deleción que otros grupos étnicos34.
Si bien la frecuencia de del22q11 fue mayor en aquellos pacientes con al menos otra anomalía mayor extracardíaca (31% vs. 18%), cabe destacar que el 65% del total de pacientes con esta deleción presentaban CCCA. El diagnóstico temprano contribuye a la prevención y a un mejor manejo de las posibles complicaciones posteriores, como inmunodeficiencia, hipocalcemia, retraso del desarrollo y del habla, trastornos del comportamiento y enfermedades psiquiátricas frecuentes en el síndrome de del22q1135. Estos signos clínicos no son evidentes al nacimiento, por lo que los resultados de este estudio son consistentes con el hallazgo de un alto porcentaje de CCCA entre los pacientes con la deleción, en donde la mediana de edad al diagnóstico fue de 2,3 meses. En conjunto, las observaciones refuerzan la necesidad de realizar el estudio molecular a niños con CCC al nacimiento.
El análisis por MLPA también evidenció un paciente con una deleción intragénica en el gen TBX1, en concordancia con varios trabajos que revelan su implicancia en CC 36,37.
En forma similar a lo ya observado9-11, el 17% de las muestras analizadas por a-CGH presentan CNV patogénicas. Todos estos desbalances están ubicados en regiones previamente descriptas en la literatura; aunque algunos pacientes muestran ciertas características clínicas diferentes, está documentado que existe expresividad variable para desbalances muy similares38. Para aquellas CNV que se clasificaron como VSI, se realizará un relevamiento periódico de la literatura con el fin de determinar si existe información adicional que permita estimar o desestimar su implicancia en la patología.
Muchas de las anomalías halladas en el a-CGH poseen un tamaño mayor a 5 Mb, superior al límite de resolución del análisis citogenético por microscopía. La mayoría de estos casos, sin embargo, corresponden a fracasos técnicos en el estudio citogenético. Si bien en Argentina, particularmente en el ámbito de la salud pública, la utilización del a-CGH resulta más costosa (a pesar de que permite estudiar una mayor cantidad de muestras en un menor tiempo y con menor cantidad de recursos humanos), estos resultados indican que su aplicación es de vital importancia en esta población de pacientes para arribar a un diagnóstico certero. De hecho, el a-CGH se utiliza en muchos países como la opción diagnóstica inicial para el estudio de las causas genéticas en pacientes con ACM39. En Argentina aún no se ha sistematizado su uso, aunque se cuenta con instituciones públicas, entre ellas el CNGM, con capacidad tecnológica y profesional para su aplicación.
Finalmente, los análisis realizados para evidenciar cambios de secuencia en el gen NKX2-5 no arrojaron la presencia de variantes (incluidas las noveles halladas) que en principio se asocien a la afección en la muestra estudiada. Sin embargo, algunas variantes que no producen cambio de aminoácidos (variantes sinónimas) podrían ejercer un efecto modulador sobre la función del gen como factor de transcripción40. En este sentido, sería interesante analizar en forma experimental la variante sinónima hallada en uno de los pacientes a los efectos de estimar si posee algún efecto biológico. Asimismo, y alternativamente, el hallazgo de variantes noveles en individuos de esta población amerita el análisis de más sujetos a fin de establecer sus posibles implicancias en la patogenia de las CC. Por otro lado, dada la cantidad de genes que ya se relacionan con la patología 15, es de esperar que el número de pacientes que presenten variantes de relevancia clínica en un gen determinado sea reducido. Si bien se ha señalado que alrededor del 5-20% de los pacientes con CC presentan variantes puntuales como causa de su afección, es sabido que ellas son más frecuentes en pacientes con antecedentes familiares o sindrómicos 41-42. Teniendo en cuenta estas consideraciones, en la actualidad se está reemplazando el enfoque hacia el estudio de varios genes en paralelo mediante la secuenciación de nueva generación (o NGS, por sus siglas en inglés).
Este estudio posee algunas limitaciones. La investigación se realizó sobre una serie de casos, y la muestra de los pacientes incorporados fue por conveniencia. Por lo tanto, no es posible extrapolar los resultados al resto de la población, tanto en lo que se refiere a las características clínicas como a las frecuencias de las anomalías genéticas detectadas. Sin embargo, cabe destacar que las muestras provinieron de centros asistenciales de diferentes jurisdicciones del país. Otra limitación del estudio fue la imposibilidad de realizar estudios de a-CGH y de variantes de secuencia en el gen NKX2-5 en un número más elevado de pacientes. En conclusión, tras el estudio de anomalías cromosómicas, desbalances genómicos y secuenciación del gen NKX2-5 en pacientes con CC, se halló la etiología de la afección en el 13% de los casos, en el marco de un estudio de pacientes con CCA y CC asociadas a otras anomalías congénitas que no había sido abordado previamente mediante este algoritmo en Argentina.
RELEVANCIA PARA POLÍTICAS E INTERVENCIONES SANITARIAS
La posibilidad de identificar la causa de la CC evita un número excesivo e innecesario de consultas médicas en busca del diagnóstico; asimismo, permite orientar el tratamiento de los pacientes y evaluar los riesgos de recurrencia para eventualmente tomar decisiones de planificación familiar. El diagnóstico temprano de afectados con CCCA que presentan del22q11 facilita un adecuado seguimiento clínico y contribuye al manejo y prevención de las posibles complicaciones posteriores. A su vez, la introducción en el ámbito público del estudio de a-CGH permite definir la causa de la CC en pacientes que, de otro modo, permanecerían sin diagnóstico.
RELEVANCIA PARA LA FORMACIÓN DE RECURSOS HUMANOS EN SALUD
El desarrollo de este trabajo contribuye a conformar un equipo multidisciplinario integrado por médicos genetistas, cardiólogos, neonatólogos, epidemiólogos, citogenetistas, bioquímicos y biólogos moleculares para la investigación de las causas de las CC; a desarrollar protocolos de estudio con la participación de diferentes profesionales de la salud; a fortalecer la capacitación profesional en la interpretación de resultados derivados de la aplicación de nuevas tecnologías genómicas; y a proveer información al equipo de salud sobre las causas genéticas de las CC.
RELEVANCIA PARA LA INVESTIGACIÓN EN SALUD
Los resultados del trabajo contribuyen a describir la presencia de alteraciones genéticas como posible causa de afección en niños de esta población con CC; a comprender los posibles mecanismos patológicos relacionados con su desarrollo; y a extender este tipo de abordajes a otras patologías de origen total o parcialmente genético.
AGRADECIMIENTOS
A las familias que aceptaron participar en este estudio y a los profesionales y técnicos de los distintos centros de salud: Lic. Noemí Buzzalino, Téc. Tania Castro, Téc. Belén Benavídez Mori, Dra. Laura Antonietti, Dra. Natalia Arros-pide, Dra. Emilia Scadizzo, Bioq. Verónica Qualina, Téc. Ezequiel Romero, Dra. Pilar Anoni, Dr. Fabián Tomasoni, Dra. Graciela Luna, Dra. María Luján Zalazar, Dra. Delfina Stremiz, Dr. Melvin Barrantes, Dr. Fernando Monti, Dra. Yamila Flores, Dra. Graciela Carballido, Dra. Viviana Heevel, Dra. Valeria Gómez, Dra. Natalia Molina, Dra. Cecilia Iraira, Dra. Claudia Cuesta, Dra. Valeria Vera, Dra. María Ángeles Vilardo, Dr. Leoncio Billordo, Dra. Jaquelin Garello, Dr. Víctor Marques, Dra. María Márquez, Dra. Mirta Raggio, Dra. Olga Mangiante, Dra. Daniela Amor, Dra. Mónica Jewtuszyk, Dra. Blanca Senra, Dra. Natalia Izzo, Dra. Mariana Brautigam, Dra. Felicitas Fumiere, Dra. Graciela Fernández, Dra. María del Carmen Arbones y Dra. Norma Cecotti.

