










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de nefrologia, dialisis y trasplante
versión On-line ISSN 2346-8548
Rev. nefrol. dial. traspl. vol.42 no.3 Ciudad Autónoma de Buenos Aires set. 2022
Artículo Original
Prevalencia de daño renal previo al diagnóstico de enfermedad de Fabry en Argentina
Prevalence of renal damage previous to the diagnosis of Fabry disease in Argentina
Sebastián Jaurretche
Norberto Antongiovanni
Fernando Perretta
1) Trasplante Renal, Pancreático y Cardíaco. Sanatorio Parque de Rosario, Rosario, Santa Fe, Argentina
2) Cátedra de Biofísica y Fisiología Humana, Escuela de Medicina, Instituto Universitario Italiano de Rosario, Rosario, Santa Fe, Argentina
3) Centro de Infusión y Estudio de Enfermedades Lisosomales, Instituto de Nefrología Clínica Pergamino, Pergamino, Buenos Aires, Argentina
4) Servicio de Terapia Intensiva, Hospital Dr. Enrique Erill de Escobar, Belén de Escobar, Buenos Aires, Argentina
Corresp ondencia: Sebastián Jaurretche ORCID: 0000-0002-1462-2703 sebastianjaurretche@hotmail.com
Recibido: 21-08-2021
Corregido: 17-11-2021
Aceptado: 23-11-2021
RESUMEN
El daño renal es una de las complicaciones mayores de la enfermedad de Fabry y su forma de presentación clínica es la proteinuria y disminución progresiva del filtrado glomerular. La frecuencia de daño renal previo al diagnóstico de enfermedad de Fabry ha sido informada en el orden del 8.7% para varones con enfermedad de Fabry "clásica", 3% para varones con enfermedad de Fabry "no clásica", 1.4% para mujeres con enfermedad de Fabry "clásica" y del 0,7% en mujeres con enfermedad de Fabry "no clásica", en pacientes de Alemania, Reino Unido y Holanda, no existiendo a la fecha información al respecto en nuestro país. Objetivo: Evaluar la frecuencia de daño renal previo al diagnóstico de la enfermedad de Fabry en Argentina. Material y métodos: Estudio epidemiológico de corte transversal realizado en tres centros de Argentina. Resultados: Se incluyeron datos de la primera consulta de 72 pacientes con enfermedad de Fabry; 44 (61,1%) pacientes presentaron daño renal previo al diagnóstico. Las variables independientes que se correlacionaron con significancia estadística a "daño renal previo" fueron la edad (p=<0,001), el genotipo (p=0,009) y el ser diagnosticado en edad adulta (p=<0,001). No se encontró correlación entre daño renal previo y las siguientes variables independientes: género (p=0,421), ser "caso índice" (p=0,139), el tipo de variante (clásica/ tardía) de la enfermedad de Fabry (p=0,107) y la actividad enzimática de la enzima lisosomal a-galactosidasa-A (p=0,916). Conclusiones: El daño renal ocurre frecuentemente previo al diagnóstico en los pacientes afectados por enfermedad de Fabry. Ante la presencia de nefropatía manifiesta por caída del filtrado glomerular estimado o proteinuria de causa desconocida, la enfermedad de Fabry debe ser considerada como diagnóstico diferencial en pacientes de ambos géneros, tanto pediátricos como adultos.
Palabras clave: enfermedad de Fabry; nefropatía; daño renal; diagnóstico temprano.
ABSTRACT
Kidney damage is one of the major complications of Fabry disease and its clinical presentation is proteinuria and a progressive decrease in glomerular filtration rate. The mfrequency of renal damage prior to the diagnosis of Fabry disease has been reported in the order of 8.7% for men with "classic" Fabry disease, 3% for men with "non-classic" Fabry disease, 1.4% for women with "classic" Fabry disease and 0.7% in women with "non-classic" Fabry disease, in patients from Germany, the United Kingdom and the Netherlands, with no information on this in our country to date. Objective: To evaluate the frequency of kidney damage prior to the diagnosis of Fabry disease in Argentina. Material and methods: Cross-sectional epidemiological study conducted in three centers in Argentina. Results: Data from the first consultation of 72 patients with Fabry disease were included; 44 (61.1%) patients had kidney damage prior to diagnosis. The independent variables that correlated with statistical significance to "previous kidney damage" were age (p=<0.001), genotype (p=0.009) and being diagnosed in adulthood (p=<0.001). No correlation was found between previous kidney damage and the following independent variables: gender (p=0.421), being an "index case" (p=0.139), type of variant (classic/late) of Fabry disease (p=0.107) and the enzymatic activity of the lysosomal enzyme a-galactosidase-A (p=0.916). Conclusions: Kidney damage frequently occurs prior to diagnosis in patients affected by Fabry disease. In the presence of manifest nephropathy due to a drop in the estimated glomerular filtration rate or proteinuria of unknown cause, Fabry disease should be considered as a differential diagnosis in patients of both genders, both pediatric and adult.
KEYWORDS: Fabry disease; nephropathy; renal damage; early diagnosis.
INTRODUCCIÓN
La enfermedad de Fabry (EF) es un error innato del metabolismo, producido por mutaciones del gen GLA, que codifica la secuencia proteica de la enzima lisosomal a-galactosidasa-A (aGalA). La actividad deficiente o ausente de aGalA resultante conduce a la progresiva acumulación de glicoesfingolípidos no catabolizados, principalmente globotriaocilceramida (Gb3) y globotriaocilesfingosina (lyso-Gb3), en plasma y diversos tejidos.(1)
El gen GLA se encuentra localizado en cromosoma X, en posición Xq22.(1) Más de 1000 variantes del gen GLA han sido descriptas, aunque algunas no tienen significado patológico (polimorfismos o variantes benignas) mientras que otras todavía permanecen sin tener un claro rol como causante de la EF (variantes de significado incierto). Tanto las variantes patogénicas como los polimorfismos y las variantes de significado incierto, deben ser consideradas en el contexto clínico de cada paciente con sospecha diagnóstica de EF al momento de decidir el inicio de tratamiento, de lo contrario, podrían recibir terapia innecesariamente algunos pacientes, y se podrían perjudicar aquellos con EF que no la reciban, (con elevados costos para el sistema de salud).(2-3) En adición al diferente significado patogénico de las variantes del gen GLA, es posible la aparición de mutaciones "de novo" (alteración genética que se presenta por primera vez en un miembro de una familia) (4) o "nuevas mutaciones" (mutación no conocida como causante de EF hasta el momento, "novel mutation").(5) Existe, además, la posibilidad de que una mutación patogénica, sea "respondedora" a chaperonas farmacológicas,(6)
una nueva alternativa terapéutica para la EF, en un grupo seleccionado de pacientes.(6) Por todo lo antedicho, es aconsejable el asesoramiento por un experto al momento de la toma de decisiones terapéuticas.(3)
Los varones con la variante "clásica" de la EF (o EF Tipo I), presentan una actividad aGalA < al 1% de lo normal, manifiestan los primeros síntomas en la niñez temprana, los cuales incluyen: i) acroparestesias y crisis de dolor neuropático en las extremidades, ii) angiokeratomas, iii) intolerancia al calor, iv) hipo o anhidrosis, v) córnea verticillata y vi) manifestaciones gastrointestinales.® Con la progresión de la enfermedad, los pacientes desarrollan enfermedad renal, cardiomiopatía y eventos cerebrovasculares a temprana edad.(1)
Por otro lado, varones con actividad residual aGalA disminuida, pero > al 1% de lo normal, no presentan los síntomas típicos de la infancia, a diferencia de los afectados por la variante "clásica". Esta variante se denomina "tardía" ("late onset", "variante del adulto", "no clásico" o EF tipo II), por su aparición más tardía y se caracteriza por el compromiso de uno o unos pocos órganos, por lo general el corazón o el riñón.(1)
A pesar de la herencia ligada al cromosoma X, las mujeres afectadas pueden presentar síntomas, aunque con gran variabilidad en su fenotipo, desde asintomáticas o con síntomas leves, hasta fenotipos "late onset" o incluso, similares a los hombres con el fenotipo "clásico" de la EF.(1,7) Dicha variabilidad clínica se produce debido a la inactivación al azar del cromosoma X (fenómeno de Lyon), lo cual resulta en un mosaicismo de expresión génica, conduciendo a una expresión diferencial de la forma mutante y funcional de la aGalA.(1, 8)
De esta manera se ha caracterizado a la población de pacientes con EF en poblaciones con Fabry "clásico" y "no clásico" tanto en varones como en mujeres como cuatro subgrupos de pacientes con manifestaciones clínicas y evolución diferente.(9)
La prevalencia estimada de la EF en la población general, ha sido reportada en 1:40000 a 1/117000 hombres,(1) aunque algunos estudios de screening neonatal han informado la posibilidad de una mayor prevalencia.(10-11) En pacientes en hemodiálisis estudios de screening informaron una prevalencia de 0,21% en varones y 0,15% en mujeres.(12) La frecuencia de daño renal previo al diagnóstico de EF, evaluada durante la primera consulta de los pacientes recientemente diagnosticados, ha sido informada en el orden del 8,7% para varones con EF "clásica", 3% para varones con EF "no clásica", 1,4% para mujeres con EF "clásica" y del 0,7% en mujeres con EF "no clásica", en pacientes de Alemania, Reino Unido y Holanda,(9) no existiendo a la fecha información al respecto en Argentina.
La detección temprana del daño renal por EF es un tema de interés entre expertos, debido a que las terapias específicas disponibles son más eficaces en estadios precoces de la nefropatía, mientras que su eficacia es limitada en etapas avanzadas (principalmente debido al desarrollo de fibrosis tisular renal durante la progresión)/13-180 o incluso, algunas alternativas terapéuticas no están indicadas en estadios avanzados de enfermedad renal crónica (ERC) por EF.(19) Existen numerosos trabajos científicos sobre la evaluación y manejo clínico de la nefropatía desde sus estadios iniciales, en los pacientes ya diagnosticados con EF;(20-25) por el contrario se encuentran disponibles escasas publicaciones destinadas a estimular al médico nefrólogo a tener en cuenta el diagnóstico de EF ante la presencia de pacientes con daño renal de causa incierta,(26-27) lo cual continúa siendo el método más redituable en términos de la ecuación costo/beneficio, en el contexto de que el daño
renal previo al diagnóstico de la EF ocurre con
frecuencia.(9)
OBJETIVOEvaluar la frecuencia de daño renal previo al diagnóstico de la EF en pacientes de Argentina.
MATERIAL Y MÉTODOSEstudio epidemiológico, de corte transversal, realizado en tres centros de Argentina, incluyendo datos de la primera consulta de pacientes con diagnóstico de EF, desde enero de 2007 hasta julio de 2021. El estudio fue aprobado por cada comité de ética local y los pacientes firmaron un consentimiento informado por escrito previo al ingreso al estudio. El presente estudio se realizó en cumplimiento de la Declaración de Helsinki.
Todos los pacientes incluidos presentaron diagnóstico confirmado de EF por manifestaciones clínicas típicas o antecedentes familiares de la enfermedad además de dosaje de actividad de aGalA más estudio mutacional por Multiplex Ligations Probe Amplification (MLPA) y secuenciación directa (Medical Genetics Laboratories-Baylor College of Medicine, Houston, TX, EE. UU.). En todos los pacientes en los cuáles se confirmó el diagnóstico de la EF se evaluó la presencia de daño renal previo, retrospectivamente, en la historia clínica de la primera consulta a cada centro.
El género de los pacientes fue considerado como variable cualitativa (varón/mujer). La edad (en años) como variable cuantitativa. El grupo etario (pediátrico/adulto) como variable cualitativa por debajo o por encima debajo de 18 años. Se consideró caso índice como variable cualitativa (SI/NO). La variante (clásico/tardío) de la EF fue determinada según la clasificación de la base de datos <http://www.dbfgp.org/dbFgp/fabry/ Mutation.html> como variable cualitativa. El dosaje de actividad aGal-A se consideró normal o disminuido por encima y por debajo de 4,0 nmol/ h/l, respectivamente, como variable cualitativa. Para evaluar daño renal se utilizó la definición de la Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline (KDIGO); se consideró daño renal la presencia de albuminuria patológica (> a 30 mg/g de creatinina) y/o FGe < a
90 ml/min/m2.(28)
Córnea verticilata fue evaluada mediante examen oftalmológico con lámpara de hendidura y fue considerada variable cualitativa (SI/NO) (i, 17) Dolor neuropático y manifestaciones gastrointestinales típicas de EF fueron evaluados por interrogatorio y su presencia o ausencia se consideró como variable cualitativa (SI/NO).(1, 17) La presencia o ausencia de compromiso del sistema nervioso central (SNC) fue considerada una variable cualitativa (SI/NO) y se definió por el antecedente de stroke y/o imágenes típicas en resonancia nuclear magnética (RMN) cerebral, aunque fueran asintomáticas.(1, 17) Se consideró arritmia cardíaca a la presencia de alteraciones electrofisiológicas típicas de la EF en electrocardiograma de 12 derivaciones como variable cualitativa (SI/NO), al igual que la presencia o ausencia de hipertrofia del ventrículo izquierdo (HVI) en ecocardiograma Doppler color y/o imágenes típicas RMN cardíaca. (1, 17) Hipertensión arterial (HTA) se consideró como variable cualitativa (SI/NO).
Estadística descriptiva: Las variables
cualitativas se expresaron como porcentajes y las variables continuas (cuantitativas) como media ± desviación estándar. Estadística inferencial: se utilizaron pruebas paramétricas o no paramétricas según el tipo de variable. Se trabajó con intervalo de confianza del 95%. Valores de p < 0,05 fueron considerados de significancia estadística. Los datos fueron procesados en una base IBM SPSS Statistics 2.0.
RESULTADOSSe incluyeron datos de la primera consulta de 72 pacientes con EF; 44 pacientes, el 61,1% de la población incluida en el estudio, presentó daño renal previo al diagnóstico de EF (Tabla 1). La Tabla 2 muestra las características demográficas y la frecuencia de daño renal previo al diagnóstico de la población en estudio. Pacientes con 9 mutaciones "clásicas" (E398X, L415P, L106R, R227Q, A292T, c.448.delG, C382Y, del
3&4exon, W81X) y 4 mutaciones "variante tardía" (M296V, R363H, R301Q, D109G) del gen GLA fueron estudiados. En el Gráfico 1 se observa la distribución de frecuencias según genotipo de los sujetos en estudio.
Tabla 1. Distribución de frecuencia de daño renal previo al diagnóstico de EF según género y variante fenotípica
| Género/Variante | Daño renal previo (n/%) | Sin daño renal previo (n/%) | Total |
| Varón/clásica | 18/64,28% | 10/35,72% | 28 |
| Mujer/clásica | 2/100% | 0/0% | 2 |
| Mujer/no clásica | 19/52,77% | 17/47,23% | 36 |
| Total | 5/83,33% | 1/16,66% | 72 |
Tabla 2. Características demográficas y frecuencia de daño renal previo al diagnóstico de la población en estudio
| Variable | n (%) | Media ± DS | Tipo |
| Género (mujeres/varones) | 42(58,3%) | - | Cualitativa |
| 30(41,7%) | (Nominal) | ||
| Edad | - | 26,26 ± 16,59 | Cuantitativa |
| Grupo etario | 24(33,3%) | - | Cualitativa |
| (pediátricos/adultos) | 48(66,7%) | (Nominal) | |
| Caso índice | 14(19,4%) | - | Cualitativa |
| (si/no) | 58(80,6%) | (Nominal) | |
| Variante Fabry | 64(88,9%) | - | Cualitativa |
| (clásica/tardía) | 8(11,1%) | (Nominal) | |
| Actividad aGalA | 34(46,6%) | - | Cualitativa |
| (normal/disminuida) | 38(52,8%) | (Nominal) | |
| Daño renal previo al diagnóstico | 44(61,1%) | - | Cualitativa |
| (si/no) | 28(38,9%) | (Nominal) |
DS: desvío estándar; aGalA: a-galactosidasa-A
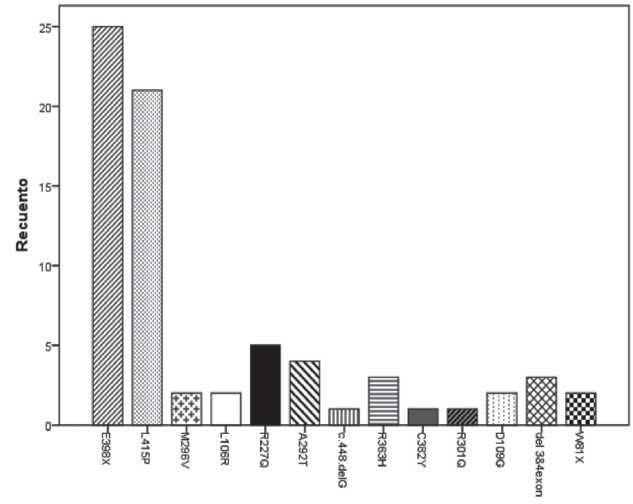
Gráfico 1. Distribución de frecuencias según genotipo de la población en estudio
Córnea verticillata fue hallada en 16 pacientes (22,2%), manifestaciones gastrointestinales en 44 (61,1°%), dolor neuropático en 26 (36,1%), angiokeratomas en 22 (30,5%), compromiso del SNC en 8 pacientes (11,1%), arritmia cardíaca en 20 pacientes (27,8%), HVI en 18 pacientes (25%)
e HTA en 19 (26,4%).
Las manifestaciones clínicas y resultados de pruebas de función renal de los pacientes, clasificados en subgrupos (con daño renal previo/ sin daño renal previo), se muestran en la Tabla 3.
Tabla 3. Manifestaciones clínicas y pruebas de función renal en pacientes con daño renal previo versus sin daño renal previo
| Variable | Sub-grupo | n (%) | Media ± DS | P |
| Córnea | Sin daño renal previo | 3 (10,71%) | - | 0,014 |
| Verticillata | Con daño renal previo | 19 (43,18%) | - | |
| Dolor | Sin daño renal previo | 6 (21,42%) | - | 0,039 |
| Neuropático | Con daño renal previo | 20 (45,45%) | - | |
| Síntomas | Sin daño renal previo | 10 (35-71%) | - | < 0,001 |
| GI | Con daño renal previo | 34 (77,27%) | - | |
| Compromiso | Sin daño renal previo | 3 (10,71%) | - | 0,933 |
| del SNC | Con daño renal previo | 5 (11,36%) | - | |
| Arritmia | Sin daño renal previo | 2 (7,14%) | - | 0,001 |
| Cardíaca | Con daño renal previo | 18 (40,90%) | - | |
| HVI | Sin daño renal previo | 6 (21,42%) | - | 0,583 |
| Con daño renal previo | 12 (27,27%) | - | ||
| HTA | Sin daño renal previo | 1 (3,57%) | < 0,001 | |
| Con daño renal previo | 18 (40,90%) | |||
| FGe | Sin daño renal previo | - | 113,83 ± 19,84 | < 0,001 |
| (ml/min/m2) | Con daño renal previo | - | 81,09 ± 43,61 | |
| Índice A/C | Sin daño renal previo Con daño renal previo | - | 8,92 ± 14,39 420,22 ± 753,47 | 0,005 |
DS: desvío estándar; Síntomas GI: síntomas gastro-intestinales; Compromiso del SNC: Compromiso del Sistema Nervioso Central; HVI: hipertrofia ventricular izquierda; HTA: hipertensión arterial; FGe: filtrado glomerular estimado; Indice A/C: índice albúmina/ creatinina en orina
Las variables independientes que se correlacionaron con significancia estadística a "daño renal previo" fueron la edad (p=<0,001, Gráfico 2), el genotipo (p=0,009, Gráfico 3) y el ser diagnosticado en edad adulta (p=<0,001, Gráfico
4). No se encontró correlación entre daño renal previo y las siguientes variables independientes: género (p=0,421), ser caso índice (p=0,139), el tipo de variante (clásica/tardía) de la EF (p=0,107) y la actividad enzimática aGalA (p=0,916).
Gráfico 2. Correlación de casos por edad entre los subgrupos "sin daño renal previo" versus "con daño renal previo"

Gráfico 3. Frecuencia de casos "sin daño renal previo" o "con daño renal previo" distribuidos por genotipo

Gráfico 4. Daño renal previo entre pacientes pediátricos y adultos

IC: intervalo de confianza; Vs: versus
DISCUSIÓN
El daño renal es una de las complicaciones mayores de la EF y su forma de presentación clínica es la proteinuria y disminución progresiva del FG.(1, 17, 29) En varones con EF "clásica", los primeros signos clínicos que indican daño renal incluyen albuminuria y proteinuria, pudiéndose desarrollar tempranamente, incluso en la niñez.(1) Cambios histológicos, potencialmente irreversibles, glomerulares, túbulointersticiales y vasculares pueden ser observados en muestras de tejido renal en niños afectados, precediendo la aparición de la albuminuria.(13, 15, 30-32) La declinación en la tasa de filtrado glomerular estimado (FGe) es poco común en edades pediátricas, pero puede ser observada en adolescentes.(33) En la edad adulta se observa el deterioro progresivo de la función renal, con un descenso anual del FGe más acentuado en varones que en mujeres afectadas, y en ambos casos más pronunciada que en la población general.(1) Sin tratamiento, los varones "clásicos" afectados evolucionan a enfermedad renal crónica terminal (ERCT), requiriendo terapias de reemplazo de función renal (TRFR) alrededor de la cuarta década de la vida.(34-35) Los pacientes con proteinuria mayor a 1 gr/24 h. y un FGe menor a 60 ml/min/m2 tienen una progresión más rápida, siendo ambos datos indicadores de peor pronóstico nefrológico.(36) En las mujeres con "variantes clásicas" el daño renal puede estar presente con frecuencia, aunque de comienzo más tardío y con una menor pendiente de progresión comparado con varones afectados.(1, 7 17) En pacientes de ambos géneros afectados por "variantes tardías" de la EF, el daño renal puede estar presente alrededor de la sexta década de la vida cuando la variante es de predominio de afectación renal o bien estar ausente en las variantes que producen afectación casi exclusivamente cardíaca.(1, 7 17)
En 2009 se informó que los pacientes afectados por EF de ambos géneros presentan una expectativa de vida disminuida respecto a la población general, debido a eventos cardiovasculares fatales.(37) En el mismo estudio se describió que un porcentaje importante de los pacientes que fallecieron de causa cardiovascular habían recibido previamente terapia dialítica. Los autores concluyeron que la mayoría de los pacientes que fallecieron presentaban severo compromiso cardíaco y renal, y que habían sido diagnosticados en forma tardía de dichas afectaciones.(37) Lo precedente puso en evidencia la relevancia y la frecuencia del compromiso renal en la EF en los pacientes de ambos sexos, habiendo sido la ERCT la principal causa de muerte en la etapa previa a la aparición de las TRFR mediante diálisis.(38)
El presente trabajo describe resultados de una extensa población de pacientes con EF, que, si bien incluye un porcentaje equitativo de varones y mujeres, no presenta una distribución uniforme de frecuencias entre pacientes "clásicos" y "tardíos", lo cual puede significar una limitante para analizar los resultados obtenidos. De todas maneras, por tratarse la EF de una enfermedad de baja prevalencia, el tamaño muestral ha permitido un análisis inferencial de los datos.
En nuestra población, un importante número de pacientes presentaban daño renal previo al diagnóstico de la EF. En los pacientes con daño renal previo se observó nefropatía más avanzada, evidenciada por menor FGe y mayor proteinuria. En este grupo de pacientes se observó además una mayor frecuencia de HTA, un importante factor de daño renal, así como de mayor progresión. Las asociaciones de daño renal previo, tanto con la mayor edad como con el diagnóstico en edad adulta son las esperadas, debido a la mayor probabilidad de riesgo de daño renal acumulado por edad, previo al diagnóstico. La correlación entre las variables daño renal previo y genotipo deben ser consideradas en el contexto de no haber tenido la muestra una distribución uniforme por genotipo. Al respecto debería considerarse esperado que los pacientes con "variantes clásicas" tengan una mayor probabilidad de presentar daño renal previo, y que esa probabilidad sea menor o nula de acuerdo con la "variante tardía" renal o cardíaca. Por último, nuestros resultados muestran que el daño renal temprano y previo al diagnóstico de EF puede ocurrir tanto en varones como en mujeres, y también está presente en la población pediátrica.
CONCLUSIONESEl daño renal ocurre frecuentemente previo al diagnóstico en los pacientes afectados por la EF. Ante la presencia de nefropatía manifiesta por caída del FGe y/o proteinuria de causa desconocida, la EF debe ser considerada como diagnóstico diferencial en pacientes de ambos géneros, tanto pediátricos como adultos.
BIBLIOGRAFÍA
1) Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30. doi: 10.1186/1750-1172-5-30.
2) Curiati MA, Aranda CS, Kyosen SO, Varela P, Pereira VG, D'Almeida V, et al. The challenge of diagnosis and indication for treatment in Fabry Disease. J Inborn Errors Metab Screen. 2017:5:e160049. doi: 10.1177/2326409816685735.
3) Jaurretche S, Antongiovanni N, Perretta F. Controversias en la indicación de tratamiento de pacientes con enfermedad de Fabry en la Argentina. RevArgMed. 2018;6(3):167-72.
4) Jaurretche S. Mutación de novo en paciente adulto joven con compromiso neurológico, cardiológico y renal. Rev Arg Med. 2016;4(8):16-8.
5) Calabrese E, Rodríguez Botta G, Rozenfeld PA. New mutation in Fabry disease: c.448delG, first phenotypic description. Mol Genet Metab. 2021;27:100708. doi: 10.1016/j.ymgmr.2021.100708.
6) Jaurretche S. Chaperonas farmacológicas. Nueva alternativa terapéutica para la nefropatía por enfermedad de Fabry en Argentina. Rev Nefrol Dial Traspl. 2020;40(1):51-61.
7) Perretta F, Antongiovanni N, Jaurretche S. Major organic involvement in women with Fabry disease in Argentina. ScientificWorldJournal. 2018;2018:6515613. doi: 10.1155/2018/6515613.
8) Dobrovolny R, Dvorakova L, Ledvinova J, Magage S, Bultas J, Lubanda JC, et al. Relationship between X-inactivation and clinical involvement in Fabry heterozygotes. Eleven novel mutations in the alpha-galactosidase A gene in the Czech and Slovak population. JMol Med (Berl). 2005;83(8):647-54. doi: 10.1007/s00109-005-0656-2.
9) Arends M, Wanner C, Hughes D, Mehta A, Oder D, Watkinson OT, et al. Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study. J Am Soc Nephrol. 2017;28(5):1631-41. doi: 10.1681/ ASN.2016090964.
10) Sawada T, Kido J, Yoshida S, Sugawara K, Momosaki K, Inoue T, et al. Newborn screening for Fabry disease in the western region of Japan. Mol Genet Metab Rep. 2020;22:100562. doi: 10.1016/j.ymgmr.2019.100562.
11) Colon C, Ortolano S, Melcon-Crespo C, Alvarez JV, Lopez-Suarez OE, Couce ML, et al. Newborn screening for Fabry disease in the north-west of Spain. Eur J Pediatr. 2017;176(8):1075-81. doi: 10.1007/ s00431-017-2950-8.
12) Doheny D, Srinivasan R, Pagant S, Chen B, Yasuda M, Desnick RJ. Fabry Disease: prevalence of affected males and heterozygotes with pathogenic GLA mutations identified by screening renal, cardiac and stroke clinics, 1995-2017. J Med Genet. 2018;55(4):261-8. doi: 10.1136/jmedgenet-2017-105080.
13) Thndel C, Bostad L, Larsen KK, Hirth A, Vikse BE, Houge G, et al. Agalsidase benefits renal histology in young patients with Fabry disease. J Am Soc Nephrol. 2013;24(1):137-48. doi: 10.1681/ASN.2012030316.
14) Weidemann F, Sánchez-Niño MD, Politei J, Oliveira JP, Wanner C, Warnock DG, et al. Fibrosis: a key feature of Fabry disease with potential therapeutic implications. Orphanet J Rare Dis. 2013;8:116. doi: 10.1186/1750-1172-8-116.
15) Skrunes R, Tiandel C, Leh S, Larsen KK, Houge G, Davidsen ES, et al. Long-term dose-dependent agalsidase effects on kidney histology in Fabry disease. Clin J Am SocNephrol. 2017;12(9):1470-9. doi: 10.2215/ CJN.01820217.
16) Ortiz A, Abiose A, Bichet DG, Cabrera G, Charrow J, Germain DP, et al. Time to treatment benefit for adult patients with Fabry disease receiving agalsidase p data from the Fabry Registry. JMed Genet. 2016;53(7):495-502. doi: 10.1136/jmedgenet-2015-103486.
17) Ortiz A, Germain DP, Desnick RJ, Politei J, Mauer M, Burlina A, et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab. 2018;123(4):416-27. doi: 10.1016/j. ymgme.2018.02.014.
18) Arends M, Wijburg FA, Wanner C, Vaz FM, van Kuilenburg ABP, Hughes DA, et al. Favourable effect of early versus late start of enzyme replacement therapy on plasma globotriaosylsphingosine levels in men with classical Fabry disease. Mol Genet Metab. 2017;121(2):157-61. doi: 10.1016/j.ymgme.2017.05.001.
19) Germain DP, Hughes DA, Nicholls K, Bichet DG, Giugliani R, Wilcox WR, et al. Treatment of Fabry's disease with the pharmacologic chaperone migalastat. N Engl J Med. 2016;375(6):545-55. doi: 10.1056/ NEJMoa1510198.
20) Najafian B, Mauer M, Hopkin RJ, Svarstad E. Renal complications of Fabry disease in children. Pediatr Nephrol. 2013;28(5):679-87. doi: 10.1007/s00467-012-2222-9.
21) Aguiar P, Azevedo O, Pinto R, Marino J, Baker R, Cardoso C, et al. New biomarkers defining a novel early stage of Fabry nephropathy: A diagnostic test study. Mol Genet Metab. 2017;121(2):162-9. doi: 10.1016/j.ymgme.2017.05.007.
22) Politei J, Alberton V, Amoreo O, Antongiovanni N, Arán MN, Barán M, et al. Clinical parameters, LysoGb3, podocyturia, and kidney biopsy in children with Fabry disease: is a correlation possible? Pediatr Nephrol. 2018;33(11):2095-101. doi: 10.1007/s00467-018-4006-3.
23) Jaurretche S, Venera G, Antongiovanni N, Perretta F, Pérez GR. Urinary excretion of microRNAs in young Fabry disease patients with mild or absent nephropathy. Open J Nephrol. 2018;8(3):71-83. doi: 10.4236/ojneph.2018.83009.
24) Riccio E, Sabbatini M, Capuano I, Pisani A. Early biomarkers of Fabry nephropathy: a review of the literature. Nephron. 2019;143(4):274-81. doi: 10.1159/000502907.
25) Feriozzi S, Rozenfeld P. Pathology and pathogenic pathways in fabry nephropathy. Clin Exp Nephrol. 2021;25(9):925-34. doi: 10.1007/s10157-021-02058-z.
26) Jaurretche SPA, Cabrera G. Evaluación pre trasplante renal en el paciente con Enfermedad de Fabry. Dial Traspl. 2016;37(2):9-17.
27) Perretta F, Antongiovanni N, Jaurretche S. Fabry Disease: Why suspect it in patients on dialysis? JOJ Uro & Nephron. 2018;5(2):555656. doi: 10.19080/ JOJUN.2018.05.555656.
28) Stevens PE, Levin A; Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline Development Work Group Members. Evaluation and management of chronic kidney disease: synopsis of the kidney disease: improving global outcomes 2012 clinical practice guideline. Ann Intern Med. 2013;158(11):825-30. doi: 10.7326/0003-4819-158-11201306040-00007.
29) Jaurretche S, Antogiovanni N, Perretta F. Prevalence of chronic kidney disease in fabry disease patients: Multicenter cross sectional study in Argentina. Mol Genet Metab Rep. 2017;12:41-3. doi: 10.1016/j. ymgmr.2017.05.007.
30) Tondel C, Bostad L, Hirth A, Svarstad E. Renal biopsy findings in children and adolescents with Fabry disease and minimal albuminuria. Am J Kidney Dis. 2008;51(5):767-76. doi: 10.1053/j.ajkd.2007.12.032.
31) Fogo AB, Bostad L, Svarstad E, Cook WJ, Moll S, Barbey F, et al.; all members of the International Study Group of Fabry Nephropathy (ISGFN). Scoring system for renal pathology in Fabry disease: report of the International Study Group of Fabry Nephropathy (ISGFN). Nephrol Dial Transplant. 2010;25(7):2168-77. doi: 10.1093/ndt/gfp528.
32) Tondel C, Kanai T, Larsen KK, Ito S, Politei JM, Warnock DG, et al. Foot process effacement is an early marker of nephropathy in young classic Fabry patients without albuminuria. Nephron. 2015;129(1):16-21. doi: 10.1159/000369309.
33) Ramaswami U, Najafian B, Schieppati A, Mauer M, Bichet DG. Assessment of renal pathology and dysfunction in children with Fabry disease. Clin J Am Soc Nephrol. 2010;5(2):365-70. doi: 10.2215/ CJN.08091109.
34) Jaurretche S, Antongiovanni N, Perretta F. Enfermedad vascular en pacientes varones con enfermedad de Fabry en hemodiálisis: estudio de cohorte retrospectivo en Argentina. Rev Nefrol Dial Traspl. 2019;39(2):101-7.
35) Schiffmann R, Warnock DG, Banikazemi M, Bultas J, Linthorst GE, Packman S, et al. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. NephrolDial Transplant. 2009;24(7):2102-11. doi: 10.1093/ndt/gfp031.
36) Wanner C, Oliveira JP, Ortiz A, Mauer M, Germain DP, Linthorst GE, et al. Prognostic indicators of renal disease progression in adults with Fabry disease: natural history data from the Fabry Registry. Clin J nAm Soc Nephrol. 2010;5(12):2220-8. doi: 10.2215/ CJN.04340510.
37) Waldek S, Patel MR, Banikazemi M, Lemay R, Lee P. Life expectancy and cause of death in males and females with Fabry disease: findings from the Fabry Registry. GenetMed. 2009;11(11):790-6. doi: 10.1097/ GIM.0b013e3181bb05bb.
38) Mehta A, Beck M, Sunder-Plassmann G (editors). Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis, 2006.

