










Servicios Personalizados
Revista

Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista argentina de reumatología
versión impresa ISSN 0327-4411versión On-line ISSN 2362-3675
Rev. argent. reumatolg. vol.28 no.1 Buenos Aires mar. 2017
ACTUALIZACIÓN
Masa ósea y fracturas en enfermedades reumatológicas autoinmunes
Bone mass and fractures in autoimmune rheumatologic diseases
María Lorena Brance1,2, María Silvia Larroudé3
1Centro de Reumatología, Rosario.
Correspondencia: E-mail: lorenabrance@gmail.com
Introducción
El remodelado óseo es un proceso continuo que ocurre a través de la vida; en condiciones fisiológicas normales, los osteoblastos están involucrados en la formación del osteoide el cual es mineralizado y reemplaza el hueso en los sitios donde el osteoclasto produjo resorción ósea para mantener la integridad esquelética. Las enfermedades reumatológicas autoinmunes (ERA) alteran este ciclo de remodelado óseo y por sí mismas afectan negativamente la masa ósea por mecanismos inflamatorios. El impacto de la inflamación sobre el hueso depende del sitio anatómico afectado, tipo de células, factores y citoquinas presentes en el sitio comprometido y de las fuerzas locales afectadas. Las citoquinas son fundamentales en la patogénesis de la pérdida ósea inducidas por la inflamación y contribuyen al desacople del ciclo de remodelado óseo.
De igual forma, los fármacos utilizados para el tratamiento de las ERA y la deficiencia de vitamina D generan un mayor riesgo de fractura. Se define como fractura por fragilidad a aquella fractura que ocurre espontáneamente, o ante trauma mínimo como caída de propia altura. La osteoporosis y la consiguiente fractura por fragilidad son una complicación importante en pacientes con enfermedades reumatológicas. Estas fracturas pueden incrementarse por el uso de glucocorticoides, la inmovilización generada por la misma enfermedad en algunos casos y por el impacto negativo de la actividad de la enfermedad de base sobre el tejido óseo. Dosis tan bajas de 2,5 mg de prednisolona o equivalente por día administrada por más de tres meses aumentan notablemente el riesgo de fracturas vertebrales1,2. Tanto las fracturas vertebrales como no vertebrales presentan una importante alteración en la calidad de vida, con una elevada morbilidad y mortalidad, y aumento de los costos del sistema de salud.
Esta actualización se focaliza en los efectos sobre la masa ósea y fracturas en pacientes con diferentes ERA, producto de la inflamación y drogas utilizadas para cada enfermedad.
Rol de la inflamación en la masa ósea en las enfermedades reumatológicas autoinmunes
El proceso inflamatorio en todas las ERA, además de la edad de los pacientes, el sexo, y la administración de glucocorticoides (GC) juega un rol importante en la desmineralización ósea. Estos cambios producidos en la masa ósea están más estudiados en pacientes con artritis reumatoidea (AR)2.
Diversas citoquinas están involucradas en el aumento del remodelado óseo y en el desarrollo de la osteoporosis, entre otros factores. La activación de los osteoclastos juega un rol principal en la desmineralización ósea y la formación de erosiones óseas. El sistema RANK/RANKL/ osteoprotegerina es uno de los principales sistemas involucrado en la fisiopatología ósea. El RANKL (ligando del receptor activador del factor nuclear kB) es un miembro de la súper familia del factor de necrosis tumoral (TNF) que es liberado por los osteoblastos y activa a los osteoclastos al unirse al RANK (receptor activador del factor nuclear kB) de los precursores osteoclásticos (Figura 1). El inhibidor natural de RANKL es la osteoprotegerina (OPG), la cual es liberada por los osteoblastos con la finalidad de controlar el proceso de resorción3. El factor estimulante de las colonias de monocitos (M-CSF) contribuye en la proliferación, la sobrevida y la diferenciación de los precursores osteoclásticos.
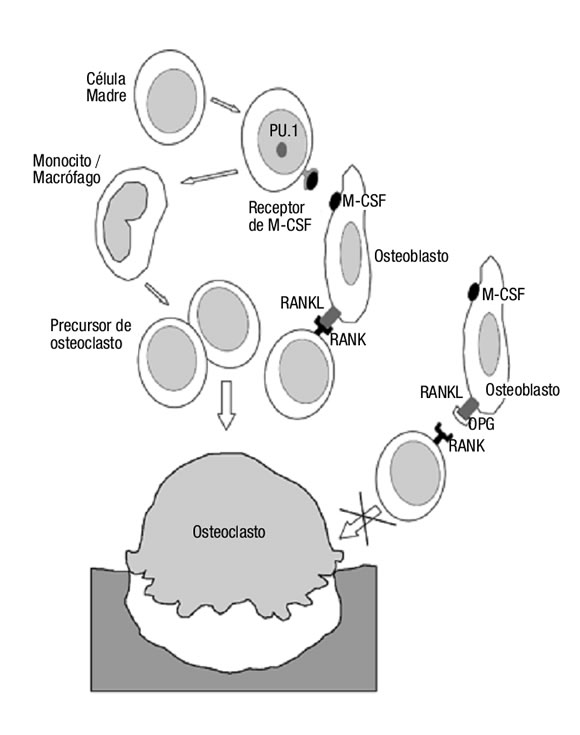
Figura 1. Diferenciación de osteoclastos a través de RANK/RANKL. OPG al unirse a RANKL inhibe la activación osteoclástica. Abreviaturas: OPG: osteoprotegerina; RANK: receptor activador del factor nuclear kB; RANKL: ligando del receptor activador del factor nuclear kB; M-CSF: factor estimulante de las colonias de monocitos; PU.1: factor de transcripción PU.1.
El control de la proliferación de las células osteoblásticas a partir de los precursores es determinante sobre la masa ósea. Los osteoblastos derivan de las células mesenquimáticas especializadas en cuya maduración tienen un rol muy importante genes específicos. Dos señales son las más importantes para la diferenciación del linaje de los osteoblastos: las BMP (bone morphogenetic protein) y el sistema canónico de wingless (Wnt). Las BMP son miembro de la superfamilia del TGF-β que incluye activinas e inhibinas. A través de diferentes vías regula la transcripción génica para la osteoblastogénesis3. Osteoprotegerina es quien estimula a la formación ósea a través de la vía canónica de las proteínas Wnt/β-catenina. Los inhibidores naturales de esta vía son la esclerostina, el dickkofs (Dkks) y la proteína secretada relacionada al frizzled (sFRP1) (Figura 2).

Figura 2. Vía de señalización intracelular Wnt/β-catenina. Abreviaturas: LRP5 y LRP6: proteína relacionada con el receptor de lipoproteína de baja densidad; APC: proteína supresora del gen de la poliposis colónica familiar; GSK-3: glucógeno sintetasa kinasa 3; TCF4: factor 4 de células T; LEF1: factor 1 de unión potenciador linfoide; Dkk1: Dickkopf 1, sFRP1: proteína 1 soluble relacionada a Frizzled, y Kremen: antagonistas extracelulares Wnt. Modificado de Canalis y cols3.
La inflamación altera el remodelado óseo a través de dos mecanismos importantes: la expresión de RANK/ RANKL y de M-CSF4.
Varias citoquinas proinflamatorias como IL-1, IL-6, IL-11, IL-15 e IL-17, producidas por los macrófagos y los linfocitos, pueden inducir la formación y la activación de los osteoclastos. El TNF-α es expresado por los linfocitos T, las células del estroma de la médula ósea y los precursores de los osteoclastos. Esta interacción entre TNF-α y su receptor produce RANKL, M-CSF e IL-1, citoquinas vinculadas a la diferenciación y activación osteoclástica5,6. En consecuencia, en las enfermedades con procesos inflamatorios, la elevada concentración de estas citoquinas produce una mayor actividad osteoclástica con mayor pérdida de masa ósea.
Masa ósea y fracturas
Artritis reumatoidea: La osteoporosis periarticular en la AR comienza a gestarse a las semanas del comienzo de la enfermedad siendo un cambio muy característico, mientras que la osteoporosis generalizada se manifesta tardíamente desde el diagnóstico siendo una causa muy importante de osteoporosis secundaria. La principal célula vinculada en la patogénesis de la AR y la destrucción ósea es el linfocito. Los linfocitos T en condiciones normales tienen la capacidad de segregar diversos tipos de citoquinas y factores, algunos que estimulan la diferenciación de los osteoclastos (IL-1, IL-6, IL-17, TNF-α, etc.) mientras que otros la inhiben (IL-4, IL-10, IL-13, IFN-γ, etc.). Sin embargo, en las enfermedades inflamatorias como la AR predomina la liberación de citoquinas proinflamatorias. Además, los linfocitos T expresan RANKL y pueden por lo tanto inducir la diferenciación de los osteoclastos. Los linfocitos T producen IL-7, la cual por un proceso independiente de RANKL puede también estimular la diferenciación osteoclástica, y al expresar TNF-α junto con el RANKL promueven la diferenciación osteoclástica. También pueden segregar IL-1, IL-6 e IL-17 todas consideradas citoquinas proresortivas, ya que estimulan la expresión de RANKL en osteoblastos y fibroblastos, permitiendo así la interacción RANK/RANKL y la diferenciación de los osteoclastos5,7.
Sin embargo, no sólo los linfocitos T son los responsables de la pérdida de masa ósea en los pacientes con AR. El proceso de inflamación generalizado que presentan estos pacientes, la inmovilidad por dolor, la atrofia muscular causada por esta última, el uso de GC y la propia actividad de la enfermedad, entre otros factores, terminan causando una disminución de la masa ósea periarticular y generalizada presentando un riesgo aumentado de fracturas vertebrales y no vertebrales. Por otro lado, el descenso de estrógenos en la mujer postmenopáusica produce un aumento de las citoquinas proresortivas y disminución de la osteoprotegerina, eventos que también conducen a incrementar la resorción ósea. La disminución de la calcemia, originada por el déficit nutricional o de la absorción tanto por déficit de vitamina D como por la acción de GC, aumentan la secreción de parathormona (PTH) la cual conduce a una mayor expresión de RANKL en osteoblastos y de forma indirecta estimulando la resorción ósea. Otro de los mecanismos de los GC es modulando factores óseos locales, los cuales disminuyen la formación ósea tanto por disminución de la diferenciación osteoblástica como por aumento de colagenasas.
Los anticuerpos anticitrulinados (ACCP), altamente específicos para AR, también están vinculados a la pérdida de hueso y dolor en pacientes con AR. Estos anticuerpos presentan diferente especificidad hacia epitopes citrulinados, y pueden desarrollarse y aparecer años antes del comienzo de la inflamación articular. Krishnamurthy y cols.8 aislaron ACCP policlonales del líquido sinovial y sangre periférica de pacientes con AR, y también aislaron ACCP monoclonales de linfocitos B de líquido sinovial también de pacientes con AR, y evaluaron entre otros puntos el desarrollo de los osteclastos incubados con y sin ACCP. Ellos hallaron que tanto los ACCP poli como monoclonales derivados de pacientes con AR pueden inducir osteoclastogénesis en monocitos humanos in vitro. Este proceso dependería tanto de la combinación de citrulinización de las proteínas intracelulares en el osteoclasto como de una disminución en la producción autócrina de IL-8. También demostraron en modelos animales que la administración endovenosa de ACCP monoclonales puede inducir pérdida de masa ósea trabecular evaluada por microtomografía computada8. En este modelo, la administración de ACCP no induce inflamación sinovial, y sí pérdida de masa ósea por bloqueo de IL-8 pero no por bloqueo de TNF, lo cual indicaría otra participación en la pérdida de masa ósea además del mecanismo inflamatorio. El estudio de Krishnamurthy y cols. sugiere fuertemente que la citrulinización dentro de los osteoclastos es importante para la osteoclastogénesis, aunque los datos no pueden identificar las moléculas citrulinadas generadas ni el modo en el cual la unión del anticuerpo media la disminución en la producción de IL-8.
Varios estudios se realizaron para evaluar la DMO en pacientes con AR y el riesgo de fractura como también evaluaron la intervención de los GC en dichos pacientes. La actividad de la AR tiene un impacto negativo en la masa ósea, efecto hallado también en pacientes premenopáusicas9.
Haugeberg y cols. describen un riesgo aumentado de dos veces para osteoporosis entre pacientes de 20 a 70 años con diagnóstico de AR10. Un estudio publicado recientemente, sobre 229 pacientes con diagnóstico de AR entre 25-55 años, halló que el 85,5% eran mujeres y de ellas el 29,9% tenía densidad mineral ósea (DMO) normal, el 37,7% osteopenia y el 40,30% tenía osteoporosis. En hombres, los hallazgos fueron similares: 15,1% tenían DMO normal, 42,4% osteopenia y 42,4% osteoporosis11. Los pacientes con AR tienen alta prevalencia de fracturas osteoporóticas, tanto vertebrales como no vertebrales12,13. Vis y cols.12 siguieron durante 5 años a mujeres postmenopáusicas con diagnóstico de AR y evaluaron la incidencia de fracturas vertebrales y no vertebrales, y hallaron que un 16% presentó una fractura no-vertebral (incidencia anual de 3,2 por cada 100 pacientes/año) y un 19% una fractura vertebral (incidencia anual de 3,7 por cada 100 pacientes/ año).
Espondilitis anquilosante (EA): La EA es una enfermedad inflamatoria crónica con compromiso del esqueleto axial caracterizada por la formación de hueso nuevo a nivel vertebral que conduce a la formación de sindesmofitos y anquilosis vertebral. También hay cambios óseos vinculados a desmineralización ósea, evidenciados por la osteopenia y la osteoporosis desarrollada. La presencia de las erosiones óseas en los ángulos vertebrales, lesión de Romanus, es expresión de activación de la vía osteoclastogénica por los cambios inflamatorios localizados.
Muchos marcadores han sido estudiados para comprender la fisiopatología de esta entidad y así enfocar un tratamiento apropiado. La formación ósea en la EA ha sido asociada con una sobre regulación de la vía Wnt. En este punto Saad y cols.14 estudiaron los niveles de esclerostina y la DMO en 30 pacientes con EA tratados con drogas anti-TNF (19 con infiximab, 9 con adalimumab y 2 con etanercept) versus controles, y hallaron bajos niveles de esclerostina en pacientes tratados con drogas anti-TNF. Otros trabajos también demostraron una disminución de los niveles de esclerostina en osteocitos de pacientes con EA15. Por otro lado, se ha hallado también expresión de TNF-α y TNF-β en biopsias realizadas de articulaciones sacroilíacas16 y bajos niveles de osteoprotegerina en pacientes con EA comparado con controles17. También hay evidencia que los niveles séricos de RANKL y el índice RANKL/OPG son significativamente más altos en pacientes con EA que en los controles18. Un trabajo que evalúo 204 pacientes con EA describió que estos pacientes presentaban niveles séricos signifcativamente más altos de Wnt y más bajos de esclerostina y RANKL comparados con los controles. A su vez hallaron que mayores niveles de proteína C reactiva (PCR) estaban asociados con niveles más bajos de esclerostina19.
Saad y cols.14 postulan que la inflamación persistente en el hueso cortical vertebral podría ser la responsable de la formación de los sindesmofitos mientras que la desmineralización en el cuerpo vertebral podría estar relacionada a mayor actividad de la vía RANKL.
Se ha hallado que la DMO de cuello femoral presenta correlación negativa con los índices de actividad de la enfermedad, con la velocidad de eritrosedimentación (VES) y la PCR18.
La prevalencia de osteoporosis en EA varía entre el 16 y el 62%20,21. Este amplio rango podría corresponder a dificultades técnicas que originan dificultades de la interpretación de los resultados, por ejemplo a que la osificación de los ligamentos, la esclerosis de los márgenes vertebrales y los sindesmofitos sobreestiman los valores de la DMO21. Esta desmineralización ósea por un lado y la proliferación ósea en determinados sitios ocurren paralelamente siendo el principal mecanismo involucrado, el proceso inflamatorio de la enfermedad de base. Contemplar este impacto fisiopatológico sobre la DMO puede explicar porqué algunos autores encuentran diferencias en la prevalencia de osteopenia, osteoporosis; otros autores, mayor afectación en columna vertebral mientras que otros encuentran alteraciones en cuello femoral. También está descripto que esta menor DMO está relacionada con la duración de la enfermedad22. La incidencia de fracturas vertebrales también varía notablemente entre el 6 y el 62%21,23. Quizás esto ocurra por una falta de exactitud en la definición entre fracturas vertebrales (la mayoría se refieren a las fracturas clínicas, manifestadas por dolor agudo en columna vertebral lo cual determina la realización de estudios y de ahí la pesquisa) versus las fracturas morfométricas (refiriéndose como tal a las fracturas asintomáticas pesquisadas por radiología). Un trabajo publicado recientemente realizado con 292 pacientes con diagnóstico de EA describe que el 20% de los pacientes presentaba fracturas vertebrales basales y un 6% desarrolló nuevas fracturas asociado a una disminución de la DMO en cadera24. Esta presencia de fracturas vertebrales a tiempo basal también fue observada en otro trabajo y relacionado con una mayor actividad de la enfermedad25. Si bien está descripto que el tratamiento con agentes anti-TNF mejoraría la DMO en pacientes con EA, en un grupo de 42 pacientes seguidos por 2 años bajo tratamiento con etanercept fue descripto un aumento del doble en la incidencia de fracturas vertebrales26. Las fracturas vertebrales en EA ocurren con el aumento de la edad, duración y severidad de la enfermedad.
Artritis psoriásica (APs): La APs afecta al hueso de formas diferentes, incluyendo tanto mecanismos de pérdida de masa ósea tales como las erosiones óseas y/o la periostitis y mecanismos involucrados en la formación ósea como la anquilosis. La prevalencia de osteoporosis llega hasta el 69% pero es controvertida, quizás por lo complejo de las interacciones mencionadas. También la presencia de fracturas -hasta el 40%- es controversial27. A pesar de esto, las fracturas por fragilidad son un problema importante en esta población. Esta alta frecuencia de fracturas vertebrales y no vertebrales también fue descripta por otros autores28.
Si bien hay estudios en APs, algunos presentan un bajo número de pacientes, no todos los estudios discriminan entre APs periférica y central, hay mucha variación en el tratamiento recibido por los pacientes, duración de la enfermedad y pocos son los estudios que estudiaron la prevalencia de fracturas principalmente vertebrales.
Pedreira y cols. estudiaron la presencia de fracturas vertebrales y no vertebrales por densitometría en 45 mujeres con APs, 52 mujeres con psoriasis y 98 mujeres sanas como grupo control. Si bien no hallaron diferencias en la DMO entre los grupos describen una mayor prevalencia de fracturas en pacientes con APs vs psoriasis (33,3% y 28,8% respectivamente; p=0,018). Las fracturas por fragilidad fueron más prevalente en los pacientes con APs y psoriasis respecto del grupo control (p=0,001). Por su parte, las fracturas vertebrales fueron más frecuentes en el grupo APs respecto del grupo con psoriasis y del grupo control (p=0,06). Es de destacar que estos pacientes con APs presentaban mayor tiempo de tratamiento con GC que las mujeres con psoriasis (9,6±22,8 y 4,8±13,2 meses, respectivamente; p=0,14)29.
Por otro lado, un trabajo que evalúo 7936 pacientes con psoriasis entre 51 y 90 años describe una asociación entre psoriasis y osteoporosis pero solo en hombres30.
Lupus eritematoso sistémico (LES): La pérdida ósea en el LES puede producirse por varios mecanismos: disminución de la movilidad, el compromiso renal, amenorrea, menopausia prematura, disminución de los niveles de andrógenos, hiperprolactinemia, inducción crónica de citoquinas, hipovitaminosis D o dependiente del tratamiento tales como el uso de GC y drogas inmunosupresoras que afectan el hueso. Gilboe y cols. describen que los pacientes con LES tienen DMO significativamente más baja (-7%) en columna lumbar, cuello femoral y cadera total comparado con pacientes controles sanos31. La frecuencia de osteoporosis en los pacientes con LES fue más alta comparada con un grupo control sano. Este grupo de trabajo comparó además pacientes con diagnóstico de LES versus pacientes con AR, describiendo que los pacientes con LES tienen similares valores de DMO en columna lumbar, cuello femoral y cadera total, comparado entre pacientes de igual edad, sexo y tiempo de duración de la enfermedad.
En pacientes lúpicos se halló menor DMO y mayor porcentaje de fracturas por fragilidad en pacientes ya expuestos a GC versus pacientes sin GC32. Un trabajo reciente que abarcó una población de 1807 pacientes lúpicos entre 1996 y 2015 describe una prevalencia de fracturas del 11,1% en pacientes con diagnóstico de LES33. Los predictores de fracturas incluyen el uso de GC, la necrosis avascular, el estado postmenopáusico y la disminución de masa ósea identificable por radiología.
Otras ERA: En cuanto a otras ERA, hay poca información en la literatura. En el Síndrome de Sjogren (SS) un grupo de trabajo, contrariamente a lo esperado, halló mayor DMO en pacientes con SS primario con acidosis tubular renal versus aquellos sin acidosis y controles34. En polimialgia reumática, se describe una asociación entre mayores niveles de VES y presencia de fracturas vertebrales en pacientes seguidos por un año, y comparados con población control35.
Los métodos habituales utilizados en los estudios previamente mencionados son la radiografía simple de columna dorsal (con foco en D8) y columna lumbar (con foco en L3) de perfil, la realización de una densitometría ósea por absorciometría de rayos X de energía dual (DXA), VFA (vertebral fracture assessment) y el laboratorio de metabolismo óseo. Otras metodologías se están utilizando en la actualidad para evaluar los cambios en la masa ósea (en investigación clínica) como lo es la tomografía computada periférica cuantitativa de alta resolución (HR-pQCT) y el trabecular bone score (TBS). En pacientes con diagnóstico de AR con esta metodología se halló que la densidad y los índices de microestructura se correlacionaron significativamente con la actividad de la enfermedad, la severidad y los niveles de citoquinas proinflamatorias36. Similares hallazgos se encontraron en pacientes con espondilitis anquilosante37. Por el contrario, en pacientes con APs, se halló que la calidad del hueso cortical, evaluada en radio distal, fue la única variable afectada correlacionando con los índices de actividad de la enfermedad38.
Acción de las drogas modificadoras de enfermedad (DMARS) en el tejido óseo
Las drogas modificadoras de la enfermedad (DMARS) han demostrado disminuir la actividad de la AR y la progresión del daño articular, como también disminuir la pérdida de masa ósea. Dolan y cols.39 evaluaron clínicamente 40 pacientes con diagnóstico de AR durante dos años en quienes determinaron marcadores bioquímicos de remodelado óseo y evaluaron la DMO al inicio del estudio y luego del primer y segundo año de seguimiento. La deoxipiridinolina urinaria, marcador de resorción, se halló significativamente aumentada en los pacientes con AR respecto del grupo control, y luego del tratamiento con metrotexate, sales de oro o sulfasalazina disminuyó significativamente respecto del valor basal, mientras que la DMO no mostró modificaciones con el tratamiento, con lo cual se interpreta que el tratamiento activo de la AR con DMARS evita la pérdida de masa ósea.
Una reciente revisión sobre los efectos de los DMARS biológicos sobre la masa ósea detalla la existencia de resultados diferentes en cuanto a su acción sobre la DMO y los niveles en los marcadores de remodelado óseo40. Estas diferencias se deben principalmente a la duración del tratamiento estudiado, y a la duración de la enfermedad en los diferentes trabajos publicados.
Está descripto que la administración de anti-TNF está asociada a un aumento de los marcadores de remodelado óseo y a una disminución de la DMO en cadera41.
El efecto de adalimumab sobre la masa ósea fue estudiado en 50 pacientes con AR activa (DAS28 ≥3,2). Luego de un año de seguimiento se halló que la DMO de columna lumbar y cuello femoral no cambió pero sí observaron un efecto sinérgico en la DMO de cuello femoral con prednisona cuando compararon adalimumab junto con prednisona (+2,5%) versus aquellos solo tratados con adalimumab (-0,7%), (p=0,015)42.
El tratamiento con infliximab en pacientes con AR mejoró la DMO en cadera total y columna lumbar luego de un año de tratamiento. Esto se acompañó de una mejoría en los niveles de los marcadores de remodelado óseo43,44. Un efecto similar está descripto en pacientes con EA. El tratamiento con infliximab luego de seis meses aumentó la DMO en columna lumbar y cadera total, junto con un aumento de los niveles de osteocalcina45. Un metaanálisis estudió el efecto de los anti-TNF en la DMO en pacientes con diagnóstico de EA. La DMO de columna lumbar aumentó un 5,1% al año de tratamiento con anti-TNF y un 8,6% luego de dos años de tratamiento46.
Los pacientes con AR tratados con etanercept también mostraron aumento de la DMO47.
El tratamiento con tocilizumab durante un año no mostró cambios en la DMO, y sí un aumento de los marcadores de formación ósea en pacientes con AR48. En pacientes refractarios a anti-TNF, se describe que tocilizumab mejora los marcadores de remodelado óseo49.
En pacientes con diagnóstico de LES, el tratamiento con rituximab luego de un año disminuyó la DMO50, mientras que en pacientes con AR produjo una disminución significativa en los niveles de deoxipiridinolina urinaria como marcador de resorción ósea, sin cambios en los marcadores de formación ósea luego de 15 meses de tratamiento51.
Glucocorticoides y masa ósea
Los glucocorticoides presentan efectos importantes en el tejido óseo y son la principal causa de osteoporosis secundaria. Los GC inducen disminución de la masa ósea, debido a sus amplios efectos en diferentes niveles del organismo: disminuyen la absorción intestinal de calcio, aumentan su excreción renal, disminuyen la liberación de estrógenos y testosterona, disminuyen la fuerza muscular, modifican factores óseos locales y alteran la actividad y diferenciación de las células óseas. La pérdida de masa ósea varía del 10 al 40% dependiendo del sitio examinado, la duración del tratamiento, la enfermedad de base y el glucocorticoide y las dosis utilizadas. El riesgo de fracturas depende de la dosis de GC. El uso de prednisona 2,5 mg/ día aumenta el riesgo relativo (RR) en 1,55, en dosis de 2,5-7,5 mg/día el RR es 2,59 y en dosis mayores el RR llega a 5,1852. Dado que la afectación es predominantemente trabecular, las fracturas vertebrales son las más frecuentes. El riesgo de fractura aumenta rápidamente luego del comienzo de la terapia con GC, y se mantiene en una fase lenta de pérdida ósea en tiempos prolongados de tratamiento. Este riesgo revierte rápidamente al discontinuar los GC, permaneciendo sin embargo un mayor riesgo relativo de fracturas en relación a la población no expuesta53,54. La prevención y tratamiento adecuado es de fundamental importancia. Diferentes guías y consensos sobre osteoporosis inducida por glucocorticoides han sido publicadas en los últimos años55,56.
Vitamina D y enfermedades reumatológicas autoinmunes
La deficiencia de vitamina D [25(OH)D] ha sido asociada con varias enfermedades autoinmunes como AR, diabetes mellitus tipo I, enfermedad inflamatoria intestinal, enfermedad mixta del tejido conectivo, enfermedad tiroidea autoinmune, esclerodermia y lupus eritematoso sistémico68,57,58,59. Se ha hipotetizado que bajos niveles de vitamina D podrían ser un factor de riesgo para el desarrollo de ERA y que niveles óptimos de 25(OH)D favorecerían la mejoría de la actividad de la enfermedad. La suplementación con vitamina D ha demostrado mejorar la enfermedad en modelos murinos de AR, diabetes mellitus tipo 1 y LES60. En pacientes con AR, la suplementación con vitamina D ha demostrado mejorar la actividad de la enfermedad61.
La presencia del receptor de vitamina D (VDR) en células del sistema inmune sugiere que la vitamina D también presenta efectos en la modulación tanto de la inmunidad innata como adaptativa62. Las células dendríticas, monocitos y macrófagos expresan 1α-hidroxilasa, la cual convierte a 25(OH)D en 1,25(OH)2 vitamina D la cual presenta efectos autocrinos y paracrinos63. Por otro lado, estudios in vitro también han demostrado que la vitamina D favorece la quimiotaxis y fagocitosis de los macrófagos, regula la liberación de ciertas citoquinas como IL-2, IL-6, inhibe la producción de IL-17 y TNF-α, reduce la proliferación y la diferenciación de células B, produciendo una disminución en la producción de inmunoglobulinas64,65,66.
El lupus eritematoso sistémico (LES) es una enfermedad autoinmune sistémica que presenta defectos en la regulación de las células T por la actividad de células B, sobreproducción de autoanticuerpos y una disminución del clearance de estos y de complejos inmunes67. Si bien algunos estudios muestran una alta prevalencia de insuficiencia y deficiencia de vitamina D en pacientes con diagnóstico de LES68, su relación con la actividad de la enfermedad no fue demostrada en todos los trabajos. Mientras algunos estudios muestran una relación recíproca entre niveles de vitamina D y actividad de la enfermedad69,70, otros no muestran asociación71.
En AR fue publicado recientemente un trabajo que evalúo 34 mujeres en comparación con 41 controles sanos61. Las pacientes con AR mostraron menores niveles de 25(OH)D (20,4±0,9 ng/ml) respecto de los controles (26,3±1,9 ng/ml). Asimismo se observó menor DMO a nivel del cuello femoral en las pacientes postmenopáusicas con AR y un mayor porcentaje de niveles subóptimos de vitamina D y una asociación inversa con la actividad de la enfermedad, la cual mejoró luego del tratamiento con vitamina D.
Datos aún no publicados mostraron menores valores de 25(OH)D en las ERA (19,17±0,66) respecto del grupo control (25,64±1,06 ng/ml). Los niveles de 25(OH) D correlacionaron negativamente con la velocidad de eritrosedimentación [r= -0,26] y con la proteína C reactiva [r= -0,27] como reactantes de fase aguda. A su vez, bajos niveles de 25(OH)D fueron encontrados en pacientes con AR que presentaban altos scores HAQ-DI y DAS 2872.
Sin lugar a dudas el conocimiento sobre su acción en la actividad de las ERA, como así el conocimiento sobre cuál es el nivel óptimo en esta acción inmunomoduladora contribuiría a una mejor respuesta del sistema inmunológico a los tratamientos seleccionados.
Conclusión
Los mecanismos patogénicos de las ERA presentan un efecto negativo sobre el tejido óseo, lo cual se expresa como una disminución en la densidad mineral ósea, un aumento de la probabilidad de fracturas vertebrales y no vertebrales. A su vez, la medicación utilizada en estos pacientes también afecta al tejido óseo. Los estudios realizados con anti-TNF evidencian un aumento de la DMO por DXA. Un mejor conocimiento de la fisiopatología de la pérdida de masa ósea será de utilidad para valorar la terapéutica más adecuada para tratar la enfermedad de base y prevenir la pérdida de masa ósea. Esto impactará en la probabilidad de sufrir una fractura y mejorará la morbimortalidad asociada.
Conflicto de interés
María Lorena Brance declara no tener conflictos de intereses. María Silvia Larroude ha recibido honorarios como orador de Genzyme, Abbie, Pfzer, Bristol, Elly Llily.
1. Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Oral corticosteroids and fracture risk: relationship to daily and cumulative doses. Rheumatology (Oxford). 2000; 39:1383-1389. [ Links ]
2. Ghazi M, Kolta S, Briot K, Fechtenbaum J, Paternotte S, Roux C. Prevalence of vertebral fractures in patients with rheumatoid arthritis: revisiting the role of glucocorticoids. Osteoporos Int. 2012;23:581-587. [ Links ]
3. Canalis E, Giustina A, Bilezikian JP. Mechanisms of Anabolic Therapies for Osteoporosis. N Engl J Med. 2007;357(9):905-916. [ Links ]
4. Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40(2):251-264. [ Links ]
5. Gillespie MT. Impact of cytokines and T lymphocytes upon osteoclast differentiation and function. Arthritis Res Ther. 2007;9(2):103. [ Links ]
6. Sims NA, Jenkins BJ, Nakamura A, et al. Interleukin-11 receptor signaling is required for normal bone remodeling. J Bone Miner Res. 2005;20(7):1093-1102. [ Links ]
7. Lacativa PG, Farias ML. Osteoporosis and inflammation. Arq Bras Endocrinol Metabol. 2010;54(2):123-132. [ Links ]
8. Krishnamurthy A, Joshua V, Haj Hensvold A, et al. Identifcation of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann Rheum Dis. 2016;75:721-729. [ Links ]
9. Fassio A, Idolazzi L, Jaber MA, Dartizio C, Viapiana O, Rossini M, Gatti D. The negative bone effects of the disease and of chronic corticosteroid treatment in premenopausal women affected by rheumatoid arthritis. Reumatismo. 2016; 68(2):65-71. [ Links ]
10. Haugeberg G, Uhlig T, Falch JA, et al. Bone mineral density and frequency of osteoporosis in female patients with rheumatoid arthritis: results from 394 patients in the Oslo County Rheumatoid Arthritis register. Arthritis Rheum. 2000;43(3):522-530. [ Links ]
11. Makhdoom A, Rahopoto MQ, Awan S, et al. Bone mineral density level by dual energy X-ray absorptiometry in rheumatoid arthritis. J Pak Med Assoc. 2017;67(1):15-19. [ Links ]
12. Vis M, Haavardsholm EA, Bøyesen P, et al. High incidence of vertebral and non-vertebral fractures in the OSTRA cohort study: a 5-year follow-up study in postmenopausal women with rheumatoid arthritis. Osteoporos Int. 2011;22:2413-2419. [ Links ]
13. Okano T, Inui K, Tada M, et al. High frequency of vertebral fracture and low bone quality in patients with rheumatoid arthritis-Results from TOMORROW study. Mod Rheumatol. 2016;2:1-7. [ Links ]
14. Saad CG, Ribeiro AC, Moraes JC, et al. Low sclerostin levels: a predictive marker of persistent inflammation in ankylosing spondylitis during anti-tumor necrosis factor therapy? Arthritis Res Ther. 2012;14(5):R216. [ Links ]
15. Appel H, Ruiz-Heiland G, Listing J, et al. Altered skeletal expression of sclerostin and its link to radiographic progression in ankylosing spondylitis. Arthritis Rheum. 2009;60:3257-3262. [ Links ]
16. François RJ, Neure L, Sieper J, Braun J. Immunohistological examination of open sacroiliac biopsies of patients with ankylosing spondylitis: detection of tumour necrosis factor alpha in two patients with early disease and transforming growth factor beta in three more advanced cases. Ann Rheum Dis. 2006;65:713-720. [ Links ]
17. Taylan A, Sari I, Akinci B, et al. Biomarkers and cytokines of bone turnover: extensive evaluation in a cohort of patients with ankylosing spondylitis. BMC Musculoskelet Disord. 2012:2;13:191. [ Links ]
18. Kim HR, Lee SH, Kim HY. Elevated serum levels of soluble receptor activator of nuclear factors-kappaB ligand (sRANKL) and reduced bone mineral density in patients with ankylosing spondylitis (AS). Rheumatology (Oxford). 2006;45(10):1197-1200. [ Links ]
19. Klingberg E, Nurkkala M, Carlsten H, Forsblad-d’Elia H. Biomarkers of bone metabolism In ankylosing spondylitis in relation to osteoproliferation and osteoporosis. Rheumatol. 2014;41(7):1349-56.
20. El Maghraoui A, Ebo’o FB, Sadni S, Majjad A, Hamza T, Mounach A. Is there a relation between pre-sarcopenia, sarcopenia, cachexia and osteoporosis in patients with ankylosing spondylitis? BMC Musculoskelet Disord. 2016; 11;17:268.
21. Leone A, Marino M, Dell’Atti C, Zecchi V, Magarelli N, Colosimo C. Spinal fractures in patients with ankylosing spondylitis. Rheumatol Int. 2016;36(10):1335-46.
22. Mitra D, Elvins DM, Speden DJ, Collins AJ. The prevalence of vertebral fractures in mild ankylosing spondylitis and their relationship to bone mineral density. Rheumatology (Oxford). 2000;39(1):85-9. [ Links ]
23. Feldtkeller E, Vosse D, Geusens P, van der Linden S. Prevalence and annual incidence of vertebral fractures in patients with ankylosing spondylitis. Rheumatol Int. 2006;26(3):234-9. [ Links ]
24. Maas F, Spoorenberg A, van der Slik BP, et al. Clinical risk factors for the presence and development of vertebral fractures in patients with ankylosing spondylitis. Arthritis Care Res (Hoboken). 2016 . doi: 10.1002/acr.22980. [ Links ]
25. Maas F, Spoorenberg A, Brouwer E, et al. Radiographic vertebral fractures develop in patients with ankylosing spondylitis during 4 years of TNF- blocking therapy. Clin Exp Rheumatol. 2016;34(2):191-199. [ Links ]
26. Van der Weijden MA, van Denderen JC, Lems WF, Nurmohamed MT, Dijkmans BA, van der Horst-Bruinsma IE. Etanercept increases bone mineral density in ankylosing spondylitis, but does not prevent vertebral fractures: results of a prospective observational cohort study. J Rheumatol. 2016;43(4):758-764. [ Links ]
27. Chandran S, Aldei A, Johnson SR, Cheung AM, Salonen D, Gladman DD. Prevalence and risk factors of low bone mineral density in psoriatic arthritis: A systematic review. Semin Arthritis Rheum. 2016;46(2):174-82. [ Links ]
28. Del Puente A, Esposito A, Costa L, et al. Fragility fractures in patients with psoriatic arthritis. J Rheumatol Suppl. 2015;93:36-9. [ Links ]
29. Pedreira PG, Pinheiro MM, Szejnfeld VL. Bone mineral density and body composition in postmenopausal women with psoriasis and psoriatic arthritis. Arthritis Res Ther. 2011;13:R16. [ Links ]
30. Dreiher J, Weitzman D, Cohen AD. Psoriasis and osteoporosis: a sex-specifc association? J Invest Dermatol. 2009;129:1643-1649. [ Links ]
31. Gilboe IM, Kvien TK, Haugeberg G, Husby G. Bone mineral density in systemic lupus erythematosus: comparison with rheumatoid arthritis and healthy controls. Ann Rheum Dis. 2000;59(2):110-115. [ Links ]
32. Sheane BJ, Gladman DD, Su J, Urowitz MB. Disease outcomes in corticosteroid-naïve patients with systemic lupus erythematosus. Arthritis Care Res (Hoboken). 2017; 69(2):252-256. [ Links ]
33. Cramarossa G, Urowitz MB, Su J, Gladman D, Touma Z. Prevalence and associated factors of low bone mass in adults with systemic lupus erythematosus. Lupus. 2016. [ Links ] pii: 0961203316664597.
34. Both T, Zillikens MC, Hoorn EJ, et al. Bone Mineral Density in Sjögren Syndrome Patients with and Without Distal Renal Tubular Acidosis. Calcif Tissue Int. 2016;98(6):573-579. [ Links ]
35. Calvo L, Pistone G, Arnone S, et al. Polymyalgia rheumatica and vertebral fractures: a 1-year pilot controlled study. Rheumatol Int. 2010;30(9):1245-1247. [ Links ]
36. Zhu TY, Griffth JF, Qin L, et al. Alterations of bone density, microstructure, and strength of the distal radius in male patients with rheumatoid arthritis: a case-control study with HR-pQCT. J Bone Miner Res. 2014;29(9):2118-2129. [ Links ]
37. Nigil Haroon N, Szabo E, Raboud JM, et al. Alterations of bone mineral density, bone microarchitecture and strength in patients with ankylosing spondylitis: a cross-sectional study using high-resolution peripheral quantitative computerized tomography and finite element analysis. Arthritis Res Ther. 2015;17:377. [ Links ]
38. Zhu TY, Griffth JF, Qin L, et al. Density, structure, and strength of the distal radius in patients with psoriatic arthritis: the role of inflammation and cardiovascular risk factors. Osteoporos Int. 2015;26(1):261-272. [ Links ]
39. Dolan AL, Moniz C, Abraha H, Pitt P. Does active treatment of rheumatoid arthritis limit disease-associated bone loss? Rheumatology (Oxford). 2002;41(9):1047-51. [ Links ]
40. Zerbini CA, Clark P, Mendez-Sanchez L, et al. Biologic therapies and bone loss in rheumatoid arthritis. Osteoporos Int. 2017;28(2):429-446. [ Links ]
41. Orsolini G, Adami G, Adami S, et al. Short-Term Effects of TNF Inhibitors on Bone Turnover Markers and Bone Mineral Density in Rheumatoid Arthritis. Calcif Tissue Int. 2016;98(6):580-5. [ Links ]
42. Wijbrandts CA, Klaasen R, Dijkgraaf MG, et al. Bone mineral density in rheumatoid arthritis patients 1 year after adalimumab therapy: arrest of bone loss. Ann Rheum Dis. 2009;68(3):373-6. [ Links ]
43. Chopin F, Garnero P, le Henanff A, et al. Long-term effects of infiximab on bone and cartilage turnover markers in patients with rheumatoid arthritis. Ann Rheum Dis. 2008;67(3):353-7. [ Links ]
44. Dischereit G, Tarner IH, Müller-Ladner U, Lange U. Infliximab improves bone metabolism and bone mineral density in rheumatoid arthritis and ankylosing spondylitis: a prospective 2-year study. Clin Rheumatol. 2013;32(3):377-81. [ Links ]
45. Allali F, Breban M, Porcher R, et al. Increase in bone mineral density of patients with spondyloarthropathy treated with anti-tumour necrosis factor. Ann Rheum Dis. 2003; 62(4): 347-349. [ Links ]
46. Haroon NN, Sriganthan J, Al Ghanim N, et al. Effect of TNF-alpha inhibitor treatment on bone mineral density in patients with ankylosing spondylitis: a systematic review and meta-analysis. Semin Arthritis Rheum. 2014;44(2):155-161. [ Links ]
47. Seriolo B, Paolino S, Sulli A, et al. Bone metabolism changes during anti-TNF-alpha therapy in patients with active rheumatoid arthritis. Ann N Y Acad Sci. 2006;1069:420-7. [ Links ]
48. Briot K, Rouanet S, Schaeverbeke T, et al. The effect of tocilizumab on bone mineral density, serum levels of Dickkopf-1 and bone remodeling markers in patients with rheumatoid arthritis. Joint Bone Spine. 2015;82(2):109-115. [ Links ]
49. Karsdal MA, Schett G, Emery P, et al. IL-6 receptor inhibition positively modulates bone balance in rheumatoid arthritis patients with an inadequate response to anti-tumor necrosis factor therapy: biochemical marker analysis of bone metabolism in the tocilizumab RADIATE study (NCT00106522). Semin Arthritis Rheum. 2012;42(2):131-139. [ Links ]
50. Mendoza Pinto C, García Carrasco M, Etchegaray Morales I, et al. Bone mineral density in systemic lupus erythematosus women one year after rituximab therapy. Lupus. 2013;22(11):1128-1134. [ Links ]
51. Hein G, Eidner T, Oelzner P, et al. Infuence of Rituximab on markers of bone remodeling in patients with rheumatoid arthritis: a prospective open-label pilot study. Rheumatol Int. 2011;31(2):269-272. [ Links ]
52. Van Staa TP, Laan RF, Barton IP, Cohen S, Reid DM, Cooper C. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum. 2003;48:3224-3229. [ Links ]
53. Adinoff AD, Hollister JR. Steroid-induced fractures and bone loss in patients with asthma. N Engl J Med 1983;309:265-268. [ Links ]
54. Van Staa TP, Leufkens HG, Cooper C. The epidemiology of corticosteroid-induced osteoporosis: a meta-analysis. Osteoporos Int 2002;13:777-787. [ Links ]
55. Brance ML, Plantalech L. Osteoporosis inducida por glucocorticoides: análisis de las guías internacionales de prevención y tratamiento. Actual Osteol. 2013;9(1):14-27. [ Links ]
56. Messina OD, Somma LF, Tamborenea MI, Castelli G, Riopedre AM, Lancioni G, Larroude MS. Guías para el diagnóstico, la prevención y el tratamiento de la osteoporosis inducida por corticoides en el adulto. Actual Osteol. 2016; 12(2):107-125. [ Links ]
57. Orbach H, Zandman-Goddard G, Amital H, et al. Novel biomarkers in autoimmune diseases: prolactin, ferritin, vitamin D, and TPA levels in autoimmune diseases. Ann N Y Acad Sci. 2007; 1109:385-340. [ Links ]
58. Vacca A, Cormier C, Piras M, Mathieu A, Kahan A, Allanore Y. Vitamin D deficiency and insufficiency in two independent cohorts of patients with systemic sclerosis. J Rheumatol. 2009;36:1924-1929. [ Links ]
59. Hajas A, Sandor J, Csathy L, et al. Vitamin D insufficiency in a large MCTD population. Autoimmun Rev. 2011;10:317-324. [ Links ]
60. Mandal M, Tripathy R, Panda AK, et al. Vitamin D levels in Indian systemic lupus erythematosus patients: association with disease activity index and interferon alpha. Arthritis Res Ther. 2014;16(1):R49. [ Links ]
61. Brance ML, Brun LR, Lioi S, Sánchez A, Abdala M, Oliveri B. Vitamin D levels and bone mass in rheumatoid arthritis. Rheumatol Int. 2015;35(3):499-505. [ Links ]
62. Pike JW, Meyer MB. The vitamin D receptor: new paradigms for the regulation of gene expression by 1,25-dihydroxyvitamin D(3). Endocrinol Metab Clin North Am. 2010;39:255-269. [ Links ]
63. Kamen DL, Tangpricha V. Vitamin D and molecular actions on the immune system: modulation of innate and autoimmunity. J Mol Med. 2010; 88:441-450. [ Links ]
64. Chen S, Sims GP, Chen XX, Gu YY, Chen S, Lipsky PE. Modulatory effects of 1,25-dihydroxyvitamin D3 on human B cell differentiation. J Immunol 2007;179:1634-1647. [ Links ]
65. Baeke F, Takiishi T, Korf H, Gysemans C, Mathieu C. Vitamin D: modulator of the immune system. Curr Opin Pharmacol. 2010;10:482-496. [ Links ]
66. Sigmundsdottir H, Pan J, Debes GF, et al. DCs metabolize sunlight-induced vitamin D3 to ‘program’ T cell attraction to the epidermal chemokine CCL27. Nat Immunol. 2007;8:285-293.
67. Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med. 2008;358:929-939. [ Links ]
68. Toloza SM, Cole DE, Gladman DD, Ibañez D, Urowitz MB. Vitamin D insufficiency in a large female SLE cohort. Lupus. 2010;19:13-19. [ Links ]
69. Amital H, Szekanecz Z, Szücs G, et al. Serum concentrations of 25-OH vitamin D in patients with systemic lupus erythematosus (SLE) are inversely related to disease activity: is it time to routinely supplement patients with SLE with vitamin D? Ann Rheum Dis. 2010;69:1155-1157. [ Links ]
70. Cutolo M, Otsa K, Paolino S, Yprus M, Veldi T, Seriolo B. Vitamin D involvement in rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis. 2009; 68:446-447. [ Links ]
71. Kim HA, Sung JM, Jeon JY, Yoon JM, Suh CH. Vitamin D may not be a good marker of disease activity in Korean patients with systemic lupus erythematosus. Rheumatol Int. 2011;31(9):1189-1194. [ Links ]
72. Brance ML, Brun LR, Larroude MS, et al. Evaluation of 25-hydroxyvitamin D levels in rheumatologic diseases. 2016 ACR-ARHP Annual Meeting [Abstract 341]. [ Links ]

