










Servicios Personalizados
Revista

Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista argentina de reumatología
versión impresa ISSN 0327-4411versión On-line ISSN 2362-3675
Rev. argent. reumatolg. vol.28 no.1 Buenos Aires mar. 2017
CASO CLÍNICO
Síndrome Klippel-Trenaunay: comunicación de dos casos
Klippel-Trenaunay syndrome: communication of two cases
R. Gontero, A. Ortiz, S. Roverano, S. Paira
Servicio de Reumatología, Hospital José María Cullen, Santa Fe.
Correspondencia: E-mail: pairasergio@hotmail.com
Resumen
El síndrome de Klippel-Trenaunay (SKT) se caracteriza por una tríada clásica, que consiste en una malformación capilar cutánea, hipertrofia del tejido blando y/u óseo en extremidades, y malformaciones venosas resultantes en venas varicosas u otras malformaciones del sistema venoso profundo. Se diagnostica mediante la presencia de cualquiera de dos de las tres características antes mencionadas. Se debe tener presente que el SKT puede presentar manifestaciones atípicas como sangrados y úlceras producidas por malformaciones vasculares en el tracto gastrointestinal, genitourinario, bazo, sistema nervioso central. El diagnóstico diferencial de una lesión vascular asociada con agrandamiento de las extremidades debe incluir otros trastornos como el síndrome de Sturge-Weber, el síndrome de Parkes-Weber y el síndrome de Proteus, entre otros. El síndrome de Klippel-Trenaunay con sus complicaciones multisistémicas requiere un enfoque multidisciplinario para un manejo integral. Se comunican dos casos que se presentan con las tres características del síndrome.
Abstract
Klippel-Trenaunay syndrome is characterized by a classic triad consisting of cutaneous capillary malformation, hypertrophy of the soft tissue and/or bone in extremities, and resulting venous malformations in varicose veins or other malformations of the venous system deep. It is diagnosed by the presence of any of two of the three characteristics mentioned above. It should be borne in mind that SKT may present atypical manifestations such as bleeding and ulcers caused by vascular malformations in the gastrointestinal tract, genitourinary tract, spleen, central nervous system. The differential diagnosis of a vascular injury associated with enlargement of the limbs should include other disorders such as Sturge-Weber syndrome, Parkes-Weber syndrome and Proteus syndrome among others. Klippel-Trenaunay syndrome with its multisystemic complications requires a multidisciplinary approach to comprehensive management. Two cases are reported that present with the three characteristics of the syndrome.
Introducción
El síndrome Klippel-Trenaunay (SKT) es una malformación compleja, que se manifiesta con una tríada característica: malformación capilar cutánea (mancha de vino de Oporto), hipertrofia del tejido blando y/u óseo en extremidades y malformaciones venosas (venas varicosas u otras malformaciones del sistema venoso profundo). Afecta a miembros superiores, inferiores y raramente la cabeza, tronco o cuello1. En la mayoría de los casos, la lesión es unilateral, pero el compromiso bilateral también ha sido descripto2. Se presentan dos casos con diagnóstico en la edad adulta y se realiza una revisión de la literatura.
Caso 1
Mujer de 39 años que consultó por lumbalgia mecánica y artralgias en articulaciones metacarpofalángicas e interfalángicas proximales. En el examen físico se observó angioma capilar en dorso de mano, muñeca y región lumbar (Figuras 1 y 2), macrodactilia en 3º y 4º dedos de mano derecha (Figura 3) y de 1º y 2º dedo en pie bilateral con sindactilia de 2º y 3º dedos de pie izquierdo (Figura 4), várices groseras en miembros inferiores (Figuras 5 y 6) y mama supernumeraria. Datos de laboratorio: hemograma, hepatograma, función renal, proteinograma por electroforesis y sedimento de orina normales. En radiografía de manos se halló engrosamiento de la cortical de metacarpianos y aumento de partes blandas en 3º y 4º dedos de mano derecha (Figura 7) y en radiografía de pies engrosamiento de la cortical de metatarsianos y falanges aumentadas de longitud (Figura 8).
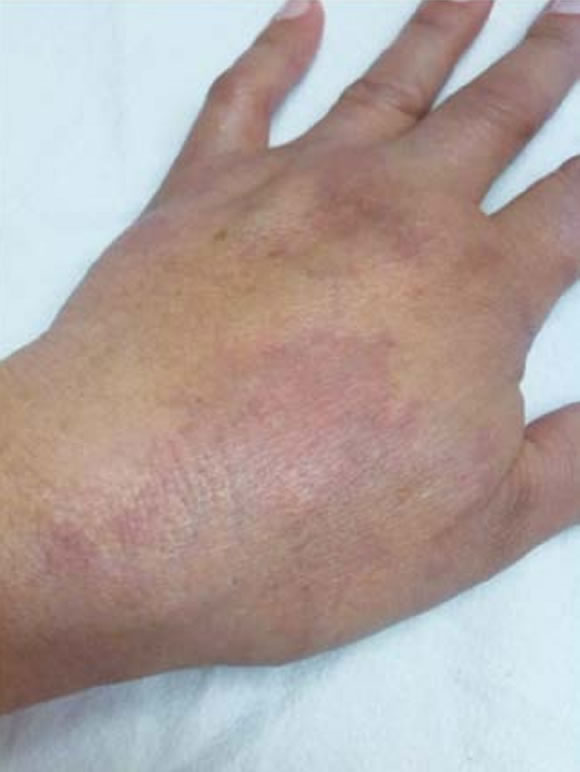
Figura 1. Mancha de vino Oporto.
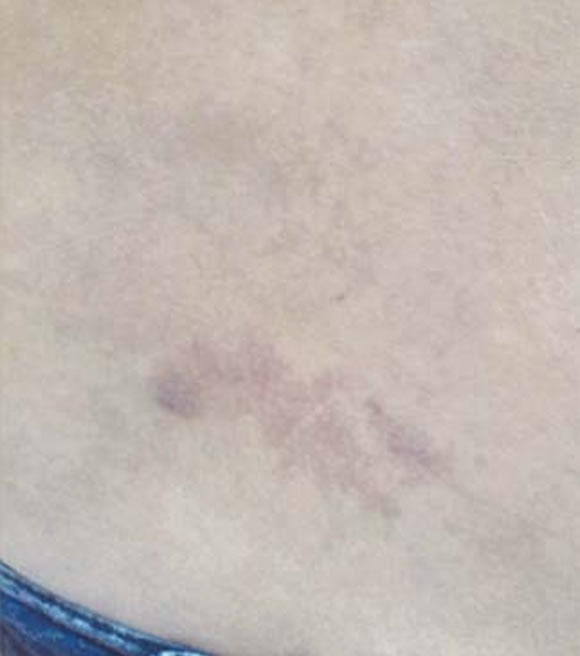
Figura 2. Mancha de vino Oporto en abdomen.

Figura 3. Macrodactilia 3º y 4º dedos mano derecha.
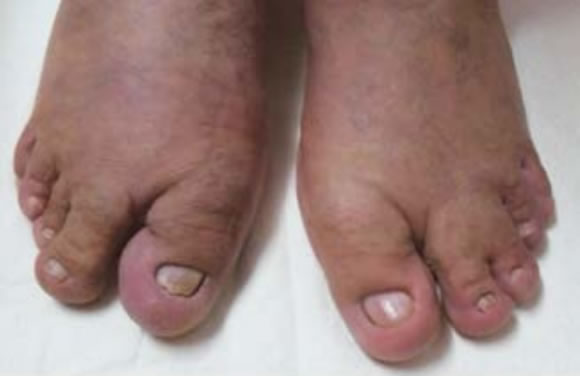
Figura 4. Macrodactilia 1º y 2º dedos bilateral y sindactilia 3º y 4º dedos pie izquierdo.
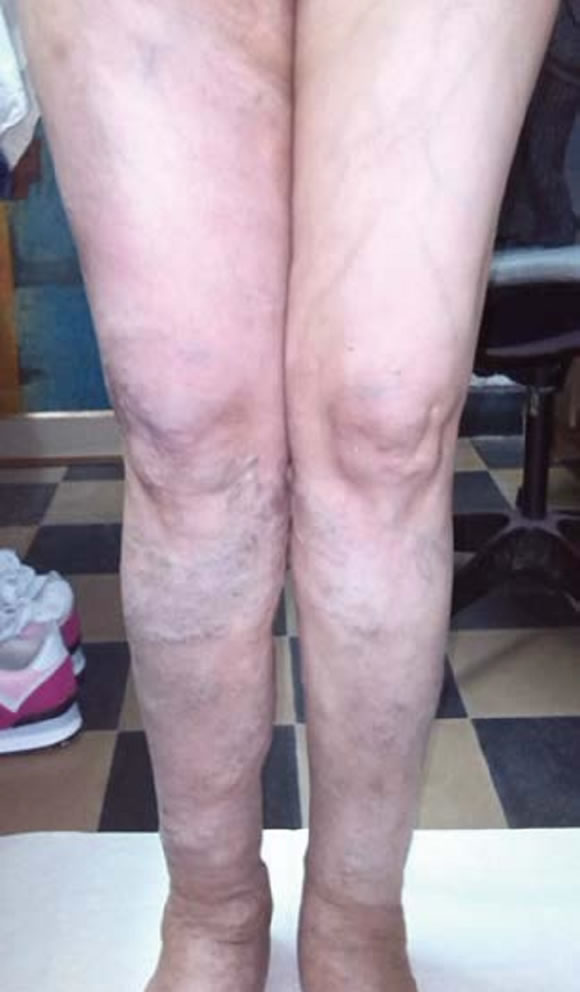
Figura 5. Várices en miembros inferiores.

Figura 6. Várices en miembros inferiores.
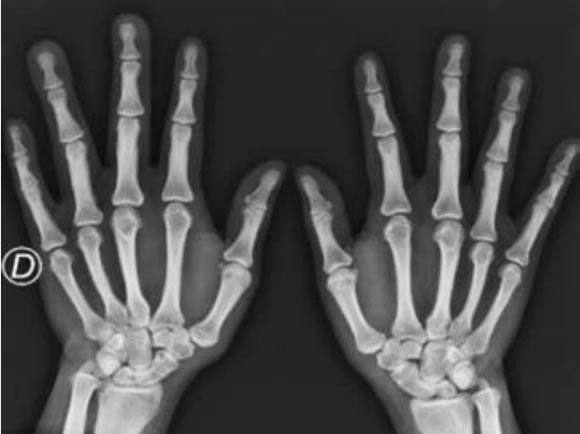
Figura 7. Aumento de partes blandas 3º y 4º dedos mano derecha. Engrosamiento de la cortical de los metacarpianos.

Figura 8. Engrosamiento de cortical de metatarsianos.
Caso 2
Mujer de 41 años con reciente diagnóstico de artritis reumatoidea, cuyo examen físico mostraba la presencia de poliartritis bilateral y simétrica de pequeñas articulaciones, malformación capilar cutánea (mancha de vino de Oporto), hipertrofia del tejido blando y várices en miembro inferior derecho (Figuras 9, 10 y 11). El laboratorio evidenció hemograma, función renal, glucemia, hepatograma, valor de sedimentación globular (VSG) y PCR normales. Factor reumatoideo (látex) positivo 48 UI/ml, antiCCP > 500 UI/ ml. FAN (HeP-2), anti-DNAn (Crithidia) y anticuerpos anti- Ro, La, Sm y RNP negativos.
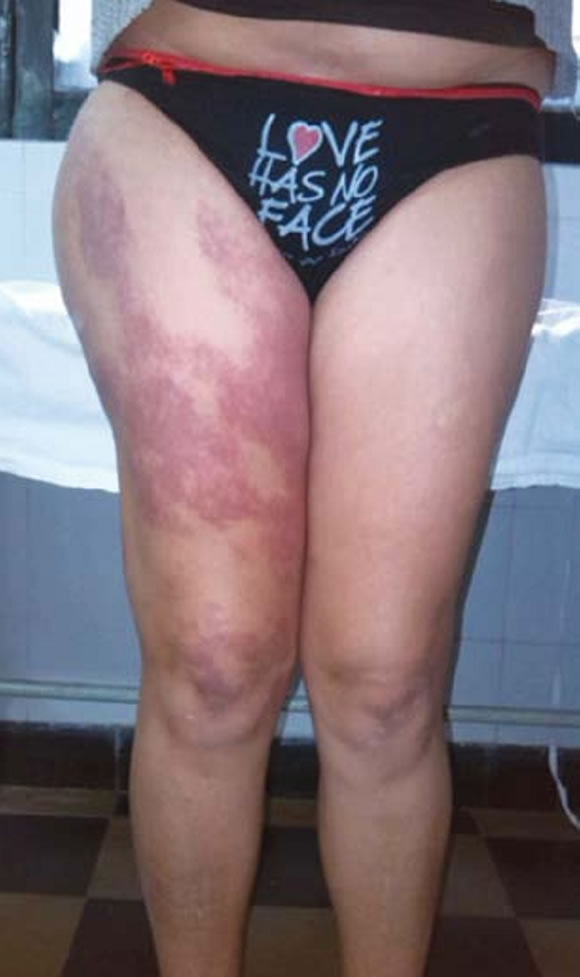
Figura 9. Mancha de vino Oporto y asimetría de miembros inferiores.

Figura 10. Mancha de vino Oporto y várices.
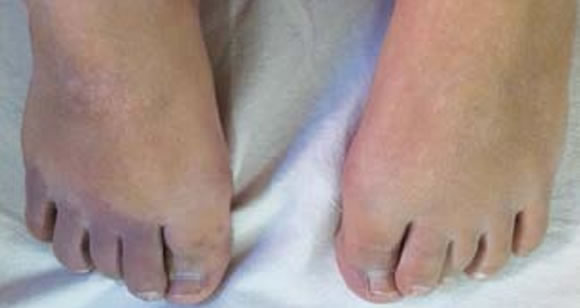
Figura 11. Sindactilia 2º y 3º dedos pie izquierdo.
Discusión
El SKT es una enfermedad vascular congénita que afecta principalmente a los vasos capilares y venas. Se define por la presencia de una tríada de signos y se diagnostica sobre dos de las tres características cardinales (mancha en vino Oporto que representa una malformación capilar, hipertrofia subyacente de huesos y tejidos blandos y venas varicosas y/o malformaciones venosas)1-4. Los casos presentados aquí mostraron las tres características del SKT: el primer caso con compromiso bilateral, lo cual es menos frecuente, y el segundo caso con compromiso unilateral, ambos sin previo diagnóstico de SKT.
La mayoría de los SKT son esporádicos aunque se han comunicado casos familiares. No hay preponderancia por sexo, raza o patrón familiar en estos casos5-7.
En una serie de 252 pacientes con SKT, el 98% presentó malformaciones capilares, 72% tenían várices o malformaciones vasculares, 67% hipertrofia de los miembros y el 63% tenía las tres características de SKT8. Se ha informado sindactilia, macrodactilia, polidactilia y displasia de cadera hasta en 29% de los pacientes. En la mayoría de los casos, la hipertrofia del tejido está limitada a las extremidades inferiores, sin embargo, también se ha informado el compromiso de cuello, tronco y extremidades superiores de manera esporádica. Otros signos y síntomas comunicados incluyen hinchazón (70%) debido a linfedema, hemorragias internas (17%), celulitis (13%) y dolor (7%)1-4. El sistema venoso marginal es la malformación característica del SKT, el cual coexiste y compite con el sistema venoso profundo pequeño que puede estar poco desarrollado. Éste muestra típicamente un flujo estancado, circunstancia que puede provocar congestión venosa asintomática, tromboflebitis superficial, trombosis venosa y tromboembolismo pulmonar. Similares complicaciones presentan las alteraciones del sistema venoso anómalo.
Con menor frecuencia puede encontrarse claudicación intermitente, úlceras varicosas, aumento de la temperatura cutánea, caída del cabello difusa, disqueratosis, alteraciones en la sudoración, producción de lágrimas y saliva. También se han observado alteraciones de la marcha y asociación con hemihipertrofia de la cara y linfangioma cutáneo, venas varicosas pulmonares y complicaciones orales6.
Siendo descripto por Klippel y Trenaunay en 1900, su etiología aún es desconocida. En principio se la atribuía a una anomalía congénita en el cordón espinal, posteriormente se pensó que las manifestaciones eran secundarias a la hipertensión venosa crónica por las alteraciones en el sistema venoso profundo. Otra hipótesis se basó en la persistencia de parte del sistema vascular embriológico, o que se trataba de un desarrollo mesodérmico generalizado anómalo9. Recientemente se han encontrado variantes genómicas en el gen AGGF1 (AnGiogenic Factor with G Patch and FHA Domains 1) que se asocian a un riesgo significativo para padecer SKT, siendo el primer gen de susceptibilidad para este síndrome. Este dato es consistente con lo hallado por Zhang y cols.10 y Baskerville P.A. y cols.9, donde la histología de las venas subcutáneas de 33 pacientes con SKT mostraba aumento en el número y en el diámetro de las vénulas en los tejidos afectados.
También fueron identificadas mutaciones en el gen PIK3CA en SKT y otros síndromes de sobrecrecimiento, el cual codifica a la 110- KD subunidad α catalática de PI3K (p110a), que al ser activada conduce a la activación de AKT y esto provoca un aumento en la proliferación celular a través de mTOR1 con crecimiento excesivo de tejido4.
Estos pacientes deben ser estudiados con ecografía doppler para evaluar la presencia de fístulas arteriovenosas, con flebografía de contraste y resonancia magnética que muestra el grado de hipertrofia tisular y el compromiso óseo y confirma las anormalidades relacionadas con el sistema linfático3,5,7.
Los diagnósticos diferenciales incluyen aquellas malformaciones que generan sobrecrecimiento y malformaciones vasculares como los síndromes de Proteus, de Sturge-Weber, de Parkes-Weber, de Maffuci, CLOVES, la anomalía fibroadiposovascular (FAVA) y Síndrome Tumor hamartoma PTEN5,6,12-14.
El SKT debe ser tratado colaborativamente por varias especialidades. El tratamiento está dirigido a las complicaciones agudas, crónicas y además la terapéutica preventiva de las anormalidades que llevaran a mayores secuelas. Muchos pacientes pueden ser manejados con medicamentos y tratamiento físico. La terapia de compresión, como el uso de una prenda/vendaje elástico, también es eficaz en pacientes que sufren de hinchazón, edema y/o dolor en las extremidades causado por hipertrofia de los miembros, estasis venosa y flebitis, pero su aplicación plantea problemas a los lactantes y niños pequeños de rápido crecimiento. El tratamiento quirúrgico está indicado cuando se vuelven excesivamente sintomáticos1,7.
Es importante destacar la morbilidad y mortalidad de esta enfermedad provocada por el tromboembolismo desencadenado por la estasis venosa, el cual puede evitarse cerrando oportunamente las venas embriológicas. Las venas marginales pueden ser obliteradas mediante embolización, fotocoagulación con láser endovenoso o se las puede remover quirúrgicamente (se la recomienda tempranamente en la infancia para evitar que alcancen un gran tamaño y por las tortuosidades). Las malformaciones linfáticas de tamaño importante provocan discapacidad, pudiendo realizarse cirugía de citoreducción. Las deformidades óseas requieren de seguimiento ortopédico; el sobrecrecimiento óseo de extremidades puede ser abordado con epificiodesis cuando ocasionan gran discrepancia. Las malformaciones capilares pueden ser tratadas con láser, las vesículas linfáticas cutáneas con escleroterapia, fotoevaporación láser con dióxido de carbono, cauterización o escisión. La evaluación hematológica es necesaria para descartar trombofilias. Las complicaciones infecciosas recibirán tratamiento oportuno con antibióticos. Estos pacientes además deben recibir acompañamiento psicológico.
Conclusión
Describimos dos pacientes con alteraciones en tamaños de extremidades y macrodactilia, sindactilia y alteraciones radiológicas óseas y de partes blandas. Sumado a la presencia de várices importantes y lesiones dérmicas, se sospechó algún síndrome que no había sido diagnosticado en los años previos. Se enfatiza la rara incidencia de esta patología, así como el bajo nivel de conocimiento acerca de esta enfermedad que resulta en un significativo retraso en el diagnóstico final.
1. H. Sreekar et al. Diverse manifestations and management options in Klippel-Trenaunay syndrome: A single centre 10-year experience. J Plast Surg Hand Surg, 2013;47:303-307. [ Links ]
2. Brodkiewicz A, Szychot E, Peregud-Pogorzelski J. Incomplete expression of klippel-Trenaunay Syndrome. Dev Period Med 2012;4:280-283. [ Links ]
3. Noel AA, Gloviczki P et al. Surgical treatment of venous malformations in Klippel-Trénaunay síndrome. J Vasc Surg 2000;32:840-7. [ Links ]
4. Vahidnezhad et al. Klippel-Trenaunay syndrome belongs to the PIK3CA-related overgrowth spectrum (PROS). Exp Dermatol 2016;25:17-19.
5. Uller W, Fishman S, Alomari A. Overgrowth syndromes with complex vascular anomalies. Semin Pediatric Surg 2014;23:208-215. [ Links ]
6. Phillips GN, Gordon DH et al. The Klippel-Trenaunay syndrome: clinical and radiological aspects. Radiology 1978;128:429-434. [ Links ]
7. Sung HM, Chung HY et al. Clinical Experience of the Klippel-Trenaunay Syndrome. Arch Plast Surg 2015;42:552-558. [ Links ]
8. Jacob A, Driscoll D, Shaughnessy W et al. Klippel-Trenaunay syndrome: spectrum and management. Mayo Clin Proc 1998;73:28-36. [ Links ]
9. Baskerville PA, Ackroyd JS et al. The etiology of the Klippel-Trenaunay syndrome. Ann. Surg. 1985; 202(5):624-627. [ Links ]
10. Zhang T, Yao Y et al. Haploinsufficiency of Klippel-Trenaunay syndrome gene Aggf1 inhibits developmental and pathological angiogenesis by inactivating PI3K and AKT and disrupts vascular integrity by activating VE-cadherin. Hum. Mol. Genet. 2016 (13). [ Links ]
11. Chen D, Li L et al. Functional characterization of Klippel-Trenaunay syndrome gene AGGF1 identifies a novel angiogenic signaling pathway for specification of vein differentiation and angiogenesis during embryogenesis. Hum Mol Genet 2013;22(5):963-976. [ Links ]
12. Edmondson AC, Kalish JM. Overgrowth Syndromes. J Pediatr Genet 2015;4:136-143. [ Links ]
13. Suárez-Amor O, Cabanillas M et al. Sturge-Weber síndrome. Ann Pediatr (Barc) 2010;73(5):299. [ Links ]
14. Banzic I, Brankovic M et al. Parkes Weber syndrome - Diagnostic and management paradigms: A systematic review. Phlebology. 2016 Aug 9. pii: 0268355516664212.

