










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista argentina de reumatología
versão impressa ISSN 0327-4411versão On-line ISSN 2362-3675
Rev. argent. reumatolg. vol.30 no.4 Buenos Aires dez. 2019
CASO CLÍNICO
Diagnóstico y manejo de la enfermedad pulmonar intersticial de la esclerosis sistémica en condiciones sanitarias desafiantes. Reporte de un caso del Hospital Universitario de Caracas
Diagnosis and management of the interstitial lung disease of systemic sclerosis in challenging public health conditions. Case report of the Caracas University Hospital
Daniel Rivas-Vargas
Hospital Universitario de Caracas, Ciudad Universitaria, Caracas
Resumen
Una mujer de 51 años presentó tumefacción en ambas manos y episodios de Raynaud trifásico de tres meses de evolución. La paciente negaba tos y disnea. En el examen físico se evidenció manos tumefactas, telangiectasias faciales y lesiones en sal y pimienta. No presentaba fibrosis de piel. La determinación de anticuerpos antinucleares y antitopoisomerasa fueron positivos. Una espirometría demostró una capacidad vital forzada (CVF) de 86% del predicho. La tomografía computarizada de tórax de alta resolución (TACAR) reveló opacidades en vidrio esmerilado bilaterales, basales y subpleurales. Estos hallazgos fueron compatibles con enfermedad pulmonar intersticial no específica (EPID) secundaria a esclerosis sistémica (ES). Además, debido a la ausencia de fibrosis de piel se diagnosticó esclerodermia sine esclerodermia. Se indicaron infusiones de ciclofosfamida endovenosa en intervalos de 4 semanas, seguido de azatioprina vía oral. Este caso ilustra que el uso de pruebas de funcionalismo pulmonar como método de detección único para la EPID puede hacer que los médicos clínicos no diagnostiquen un número representativo de pacientes, y que además, la ausencia de síntomas respiratorios no descarta la enfermedad pulmonar en pacientes con CVF normal. El pronóstico sombrío de la EPID es una indicación para la búsqueda sistémica y la realización un diagnóstico precoz.
Palabras clave: Esclerosis sistémica; enfermedad pulmonar; Caracas.
Abstract
A 51-year-old woman presented with swelling in both hands and a 3-months history of triphasic Raynaud phenomenon. She denied cough and dyspnea. The physical examination was notable for swollen hands, facial telangiectasia and salt and pepper lesions. She had no skin fibrosis. The determination of antinuclear antibodies and antitopoisomerase were positive. A spirometry demonstrated a forced vital capacity of 86% of the predicted. High resolution chest computed tomography revealed bilateral, basal and subpleural ground glass opacities. These findings were consistent with the diagnosis of Scleroderma-Related Interstitial Lung Disease. Moreover, due to the absence of skin fibrosis, a diagnosis of systemic sclerosis sine scleroderma was made. Infusions of endovenous cyclophosphamide were indicated at 4-week intervals, followed by oral azathioprine. This case showed that using PFT as the single screening method for SSc-ILD may cause clinicians to miss a significant number of patients and that the absence of pulmonary symptoms does not exclude lung disease in patients with normal FVC. The poor prognosis of SSc-ILD is an indication of systematic screening for early diagnosis.
Key words: Systemic Sclerosis; Lung disease; Caracas.
Descripción del caso
Una mujer de 51 años refirió presentar tumefacción en ambas manos de 1 mes de evolución. La paciente estaba asintomática hasta hace tres meses, cuando presentó episodios de fenómeno de Raynaud trifásico. La paciente negaba tos, disnea y síntomas de reflujo gastroesofágico. En el examen físico se evidenció telangiectasias faciales, edema leve indoloro con piel brillante en ambas manos congruente con manos tumefactas (manos puffy) (Fig. 1), y una mácula despigmentada que abarcaba el área interescapular que respetaba las áreas perifoliculares, compatibles con lesiones en “sal y pimienta”. No presentaba microstomía, úlceras digitales, engrosamiento de piel ni calcinosis.

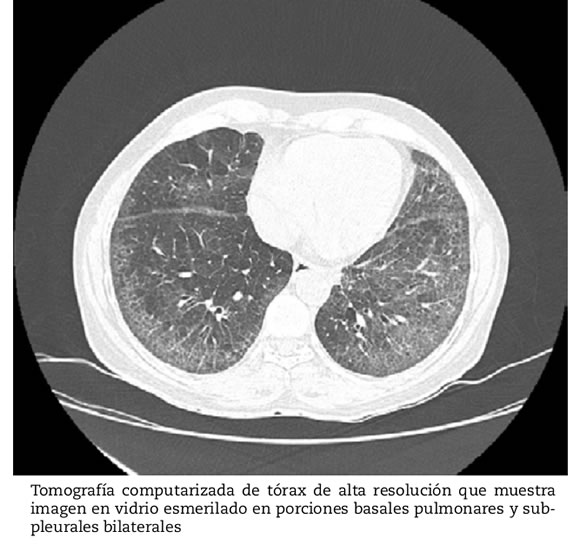
La determinación de anticuerpos antinucleares por inmunofluorescencia indirecta fueron positivos a títulos de 1: 160 con patrón nucleolar, y el ensayo inmunoabsorbente ligado a enzimas en búsqueda de anticuerpos antitopoisomerasa fue positivo. Una espirometría demostró una capacidad vital forzada (CVF) de 86% del predicho, y en la caminata de seis minutos recorrió 425 metros en el primer intento y 446 metros en el segundo intento. Se realizó una TACAR de tórax que reveló opacidades en vidrio esmerilado bilaterales, basales y subpleurales, compatibles con neumonía intersticial no específica EPID (Fig. 2).
Con estos elementos se realizó el diagnóstico de enfermedad pulmonar intersticial secundaria a ES. A pesar de la ausencia de esclerodactilia y fibrosis dérmica (modified Rodnan Skin Score de 0 pts) se clasifica como una esclerodermia sine esclerodermia. Se prescribió iniciar tratamiento con infusiones endovenosas de ciclofosfamida 600mg/m2 a intervalos de 4 semanas durante 6 meses seguido de azatioprina oral 2.5 mg/kg diario como mantenimiento.
El edema de manos de la paciente mejoró tras culminar las primeras infusiones de ciclofosfamida. Tras culminar las lesiones se iniciará azatioprina vía oral. El paciente se mantendrá bajo control con pruebas de función pulmonar cada 6 meses y control de imagen pulmonar.
Discusión del caso
La ES, es una enfermedad sistémica caracterizada por daño microvascular endotelial, inflamación y autoinmunidad que desencadena la fibrosis de piel y órganos internos.1 El grado de fibrosis, y la afección de orgánica generan fenotipos con heterogeneidad clínica y pronóstico variable. Existen dos subtipos claramente diferenciados basados en la extensión de la fibrosis de piel: si el compromiso es distal a codos y rodillas se denomina esclerodermia limitada, mientras que si el compromiso es proximal recibe el nombre de esclerodermia difusa.2 Sin embargo, al menos un 5% de casos no pueden clasificarse en estas categorías; tal es el caso de pacientes con fenómeno de Raynaud en conjunción con otros signos característicos como úlceras digitales, hipertensión pulmonar, neumopatía intersticial o autoanticuerpos específicos, pero en ausencia de fibrosis de piel.3 Este subconjunto ha recibido el nombre de esclerosis sistémica sine esclerodermia, y algunos autores han planteado una subclasificación de acuerdo al orden de aparición de la fibrosis de piel y de órganos.4 Por otro lado debido a que gran parte de los casos de ES sine esclerodermia terminan por diferenciarse en alguno de los subconjuntos establecidos, es considerada como una forma incompleta de la enfermedad y no es considerada un subgrupo aparte.3
El sello de la ES es la fibrosis, que produce las manifestaciones clínicas como la enfermedad pulmonar intersticial, la cual está presente en el 80% de los pacientes y es una de las principales causas de mortalidad.2 El compromiso pulmonar en esclerodermia suele ser de inicio insidioso y asintomático, pudiéndose detectar en esta etapa sólo a través del reconocimiento temprano de la ES que motiva la realización oportuna de la TACAR. La EPID es el patrón más frecuentemente descrito, se caracteriza por la opacidad en segmentos basales y subpleurales bilaterales “en vidrio esmerilado”, que traduce alveolitis activa5 y es susceptible de reversión con el tratamiento inmunosupresor oportuno. La6 realización de espirometría durante la primera visita médica no es lo suficientemente sensible ni específico para determinar la EPID temprana7,8, por lo tanto, la TACAR debe realizarse de forma sistémica en el momento del diagnóstico de esclerosis sistémica, junto a las demás herramientas, como la prueba de capacidad de difusión de monóxido de carbono y la caminata de 6 minutos.9
En el presente caso, la paciente no presentaba síntomas respiratorios, y la EPID se diagnosticó con base en los signos característicos de la ES, que indujo la realización oportuna de la TACAR. Esto demuestra que el uso de pruebas de funcionalismo pulmonar como método de detección único para EPID puede resultar en que los médicos clínicos no diagnostiquen un número representativo de pacientes,7 y que además, la ausencia de síntomas respiratorios no descarta la enfermedad pulmonar aún en presencia de una CVF normal.3 El pronóstico sombrío de la EPID en la ES es una indicación para la búsqueda sistémica y el diagnóstico precoz.10,11 Otra herramienta diagnóstica potencial es la ecografía pulmonar.12
Conflicto de intereses
Ninguno
Fondos
El autor declara no haber recibido apoyo financiero durante la realización de la investigación.
El caso cumplió con los lineamientos de la Declaración de Helsinki y la aprobación del Comité de Ética del Hospital Universitario de Caracas. Previa inclusión del paciente en el estudio se le informó sobre los objetivos del mismo, así como su carácter no intervencionista y no perjudicial, habiéndosele solicitado la firma de su consentimiento informado previamente.
1. Denton CP, Khanna D. Systemic sclerosis. Lancet. 2017;390:1685-1699. [ Links ]
2. Schoenfeld S, Castelino F. Interstitial Lung Disease in Scleroderma. Rheum Dis Clin North Am. 2015;41:237-248. [ Links ]
3. Diab S, Dostrovsky N, Hudson M, Tatibouet S, Fritzler MJ, Baron M, et al. Systemic sclerosis sine scleroderma: a multicenter study of 1417 subjects. J Rheumatol. 2014;41:2179-85. [ Links ]
4. Kucharz EJ, Kopec-Medrek M. Systemic sclerosis sine scleroderma. Adv Clin Exp Med. 2017;26:875-880. [ Links ]
5. Bussone G, Mouthon L. Interstitial lung disease in systemic sclerosis. Autoimmun Rev. 2011;10:248-55. [ Links ]
6. White B, Moore WC, Wigley FM, Xiao HQ, Wise RA. Cyclophosphamide is associated with pulmonary function and survival benefit in patients with scleroderma and alveolitis. Ann Intern Med. 2000;132:947-54. [ Links ]
7. Suliman YA, Dobrota R, Huscher D, Nguyen-Kim TD, Maurer B, Jordan S, et al. Pulmonary Function Tests: High Rate of False-Negative Results in the Early Detection and Screening of Scleroderma-Related Interstitial Lung Disease. Arthritis Rheumatol. 2015;67:3256-61. [ Links ]
8. Showalter K, Hoffmann A, Rouleau G, Aaby D, Lee J, Richardson C, et al. Performance of Forced Vital Capacity and Lung Diffusion Cutpoints for Associated Radiographic Interstitial Lung Disease in Systemic Sclerosis. J Rheumatol. 2018;45:1572-1576. [ Links ]
9. Chowaniec M, Skoczynska M, Sokolik R, Wiland P. Interstitial lung disease in systemic sclerosis: challenges in early diagnosis and management. Reumatologia. 2018;56:249-254. [ Links ]
10. Strickland G, Pauling J, Cavill C, Shaddick G, McHugh N. Mortality in systemic sclerosis - a single centre study from the UK. Clin Rheumatol. 2013;32:1533-9. [ Links ]
11. Simeón-Aznar CP, Fonollosa-Plá V, Tolosa-Vilella C, Espinosa-Garriga G, Campillo-Grau M, Ramos-Casals M, et al. Registry of the Spanish Network for Systemic Sclerosis: Survival, Prognostic Factors, and Causes of Death. Medicine (Baltimore). 2015;94:e1728. [ Links ]
12. Tardella M, Di Carlo M, Carotti M, Filippucci E, Grassi W, Salaffi F. Ultrasound B-lines in the evaluation of interstitial lung disease in patients with systemic sclerosis: Cut-off point definition for the presence of significant pulmonary fibrosis. Medicine (Baltimore). 2018;97:e0566. [ Links ]

