










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista argentina de reumatología
versão impressa ISSN 0327-4411versão On-line ISSN 2362-3675
Rev. argent. reumatolg. vol.31 no.2 Buenos Aires jun. 2020
CASOS CLÍNICOS
Policondritis recidivante como fenómeno paraneoplásico en paciente con síndrome mielodisplásico
Relapsing Polychondritis as paraneoplastic phenomenon in patient with Myelodysplastic Syndrome
Nadia Riscanevo, Diego Baenas, Janet Flores, Francisco Caeiro, Verónica Saurit, Alejandro Alvarellos, Juan Pablo Pirola, Julieta Olmedo, Gastón Caeiro
Servicio de Reumatología, Hospital Privado Universitario de Córdoba (Argentina)
Correspondencia: Servicio de Reumatología, Hospital Privado Universitario de Córdoba, Naciones Unidas 346 (X5016KEH) Córdoba, naclaurisca@hotmail.com
Resumen
Los síndromes mielodisplásicos son un grupo heterogéneo de enfermedades hematológicas, caracterizadas por hematopoyesis ineficaz con riesgo de progresión a leucemia mieloide aguda. Pueden asociarse a manifestaciones autoinmunes en un 10-30% de los pacientes, apareciendo antes, durante o luego del diagnóstico del trastorno hematológico. La prevalencia de policondritis recidivante como fenómeno paraneoplásico es de 0,7-5,4%, presentándose de forma simultánea en la mayoría de los casos. Otros procesos autoinmunes asociados incluyen: vasculitis sistémica, poliartritis seronegativa, dermatosis neutrofílica, citopenias inmunomediadas, presencia de autoanticuerpos y crioglobulinemia. Reportamos el caso de una mujer de 60 años, sin antecedentes patológicos previos, que presentó un cuadro de policondritis recidivante y vasculitis sistémica asociadas a síndrome mielodisplásico.
Palabras clave: Síndrome mielodisplásico; policondritis recidivante; síndrome paraneoplásico
Abstract
Myelodysplastic syndromes are a heterogeneous group of hematological diseases, characterized by ineffective hematopoiesis with risk of progression to acute myeloid leukemia. They can be associated to autoimmune manifestations in 10-30% of patients, appearing before, during or after the diagnosis of the hematological disorder. The prevalence of relapsing polychondritis as a paraneoplastic phenomenon is 0.7-5.4%, occurring simultaneously in the majority of cases. Other associated autoimmune processes include: systemic vasculitis, seronegative polyarthritis, neutrophilic dermatosis, immunomediated cytopenias, presence of autoantibodies and cryoglobulinemia. We report the case of a 60-year-old woman, with no previous medical history, who presented with recurrent polychondritis and systemic vasculitis associated with myelodysplasia.
Key words: Myelodysplastic syndromes; relapsing polychondritis; paraneoplastic syndromes
Introducción
Los síndromes mielodisplásicos (SMD) son un grupo heterogéneo de enfermedades clonales de las células hematopoyéticas pluripotentes, caracterizadas por la presencia de alteraciones morfológicas en estas células, citopenias y riesgo de progresión a leucemia mieloide aguda.1 Las manifestaciones autoinmunes (MA) asociados con mayor frecuencia son: vasculitis leucocitoclástica, poliartritis seronegativa, dermatosis neutrofílica y pioderma gangrenoso.2,3 En menor medida se han reportado casos de vasculitis sistémica (símil Granulomatosis con poliangeítis o Behçet), policondritis recidivante (PR), glomerulonefritis, enfermedad de Crohn, citopenias inmunomediadas y anormalidades serológicas como la presencia de ANA, ANCA, factor reumatoideo y crioglobulinemia.2,3
Presentamos el caso de una mujer de 60 años con SMD que presentó PR y vasculitis como fenómenos paraneoplásicos y evolución a leucemia mieloide aguda.
Caso clínico:
Mujer de 60 años, sin antecedentes patológicos previos, consultó por fiebre de 39°C, hipoacusia derecha, otalgia y cefalea frontal. Inicialmente el cuadro fue interpretado como sinusitis y se indicó tratamiento antimicrobiano sin respuesta, por lo que se solicitaron estudios complementarios. En los análisis sanguíneos se evidenció anemia normocítica-normocrómica, policromatofilia, células diana, punteado basófilo, leucocitosis con linfocitos atípicos, mielocitos, metamielocitos, y eritrosedimentación elevada. Se realizó tomografía de senos paranasales en la que se observó compromiso de senos esfenoidales, maxilar derecho y celdillas mastoideas. Los hemocultivos para gérmenes comunes y hongos fueron negativos. Se realizó una punción aspiración de médula ósea que demostró signos compatibles con anemia refractaria con exceso de blastos (AREB II) y 14% de blastos mieloides (Figura 1.e). En el cariotipo se observó monosomía 7 y anomalías numéricas y estructurales agregadas en mosaico, con detección de anomalía 5q 33 - q 34. Se inició tratamiento para el SMD con azacitidina 75mg/m2 por 3 ciclos.
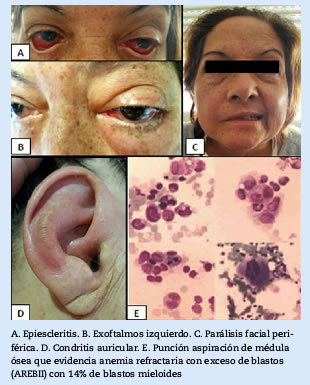
Durante su evolución la paciente agregó hipoacusia bilateral con pruebas de Rinne, Weber y audiometría compatible con alteración sensorial y de conducción. Posteriormente presentó irritación y dolor ocular bilateral intenso, que evolucionó con exoftalmo izquierdo y diplopía. Presentó mejoría parcial con prednisona 40 mg/día, pero con ulterior recaída ocular a los 10 días, sumado a parálisis facial periférica, condritis nasal y de pabellón auricular derecho. En la resonancia magnética nuclear de órbitas se evidenció infiltración de los músculos oculares extrínsecos. Se realizaron tres pulsos de metilprednisolona de 500 mg y 750 mg de ciclofosfamida con mejoría temporal de los síntomas.
Treinta días después se presentó con síndrome febril en ausencia de signos sugestivos de infección y lesiones vasculíticas periungueales (lesiones de “Bywater”), mononeuritis múltiple con afectación del III y VII par craneal, y compromiso del ciático-poplíteo izquierdo. En el laboratorio inmunológico no se evidenciaron nuevas alteraciones significativas (ANA, ENA, factor reumatoideo, crioglobulinas, antifosfolípidos y ANCA negativos, complementemia normal). Se indicaron nuevamente pulsos de metipredisolona y rituximab 1 gramo, pero la paciente persistió con pancitopenia y fiebre. Debido a la falta de respuesta al tratamiento inmunosupresor, se sospechó progresión de SMD. Se realizó nueva punción y biopsia de médula ósea en las que se evidenció evolución a leucemia mieloide aguda (blastos mieloides 34%). Por este motivo se inició tratamiento quimioterápico de inducción con daunorrubicina y citarabina, con respuesta inicial favorable de las manifestaciones autoinmunes. Un mes después desarrolló cuadro de neutropenia febril sin aislamientos microbiológicos y progresión de enfermedad de base, falleciendo a los 5 meses desde el inicio de la enfermedad. Las principales MA se muestran en la Figura 1.
Discusión
La relación entre SMD y MA ha sido estudiada en numerosas series de casos.2-5 La incidencia informada de estas manifestaciones en pacientes con SMD varía entre 10% en estudios retrospectivos y un 18.5% en un estudio prospectivo de 4 años.2,6,7
Esta asociación entre MA y SMD podría ser consecuencia de una predisposición genética común, y no existe una cronología predecible entre los dos procesos.8
En nuestro caso, la paciente debutó con MA sugestivas de vasculitis (sinusitis, hipoacusia y fiebre en ausencia de aislamientos microbiológicos) y se detectaron citopenias en la analítica general, lo cual permitió el posterior diagnóstico de SMD. A pesar del tratamiento de base del trastorno hematopoyético, la paciente presentó numerosas MA a lo largo de su evolución, cumpliendo con los criterios diagnósticos de McAdam9 de PR debido a la presencia de condritis auricular, condritis nasal, epiescleritis y disfunción coclear con respuesta transitoria a esteroides.
La PR es una enfermedad sistémica infrecuente, caracterizada fundamentalmente por inflamación recurrente y destrucción de tejidos cartilaginosos. Afecta principalmente pabellón auricular, cartílago nasal, articulaciones periféricas y árbol traqueobronquial, pero puede comprometer otras estructuras ricas en proteoglicanos como el ojo (escleritis y uveítis), la piel, oído interno, vasos sanguíneos y miocardio. La condritis auricular es la afección más frecuente (78 % de los casos) y puede presentarse como eritema marcado e incremento del volumen en el pabellón auricular, limitado a su región cartilaginosa, de forma localizada o difusa, siendo generalmente bilateral.9
La hipoacusia puede presentarse por estenosis del conducto auditivo externo secundaria a inflamación, como también a otitis media y obstrucción de las trompas de Eustaquio por condritis del segmento nasofaríngeo. Se postula que los trastornos neurosensoriales cocleares o vestibulares podrían deberse a vasculitis de la arteria auditiva interna o de sus ramas vestibular o coclear.9
El compromiso ocular se ha observado en hasta el 60% de los pacientes. La manifestación más frecuente es la epiescleritis, y en ocasiones escleritis o uveítis. Se han reportado también conjuntivitis, blefaritis, queratitis, iritis, perforación corneal, pseudotumor orbitario, parálisis de músculos extraoculares y neuritis óptica.9
El síndrome febril prolongado también podría ser una manifestación de la PR. En su evolución, nuestra paciente presentó fiebre en numerosas oportunidades, en ausencia de aislamientos infecciosos, y la mayoría de las veces en el contexto de MA.
Se ha descripto que la alteración inmunológica en SMD podría deberse a la capacidad del clon neoplásico para transformar el microambiente de la médula ósea y provocar liberación de citoquinas inflamatorias y pro leucémicas.6 Las células mieloides supresoras secretan IL-10 y TGF-Beta, y se genera un disbalance entre citocinas proinflamatorias y mielosupresoras, con activación de monocitos, células dendríticas, macrófagos y neutrófilos que se expanden en la médula ósea y gatillan la respuesta inmune.10
El tratamiento de la PR sigue siendo empírico y tiene por objetivo reducir la frecuencia y la intensidad de las exacerbaciones y prevenir el desarrollo de daño irreversible. Los glucocorticoides son utilizados en la gran mayoría de los pacientes ya que a menudo son notablemente eficaces. Se usan para tratar formas graves de la enfermedad como en caso de compromiso oftalmológico severo, afectación laríngea, traqueal o bronquial, vasculitis sistémica entre otros. Se puede realizar terapia combinada con metotrexato, azatioprina, ciclofosfamida y ciclosporina A, para ahorrar corticoides o en caso de intolerancia o resistencia. Se han reportado casos de buena respuesta a anti-TNF en pacientes refractarios a los tratamientos previos, y reportes aislados de eficacia de anti IL-1 en pacientes que fallaron a anti-TNF.9,11 La respuesta favorable a rituximab ha sido ampliamente evaluada en vasculitis sistémicas pero los resultados en policondritis recidivante son escasos y controvertidos.11,12 En una serie retrospectiva de nueve casos, dos pacientes presentaron mejoría parcial, 4 se mantuvieron estables, y 3 empeoraron a pesar del tratamiento.12
La progresión de los síntomas y signos de PR en nuestra paciente durante las últimas 2 internaciones, junto a la refractariedad al tratamiento hematológico y reumatológico, sugirieron la posibilidad de progresión de su SMD. Por este motivo, se realizó una nueva punción y biopsia medular, confirmándose la progresión a leucemia. Se ha observado un rápido deterioro clínico y elevada mortalidad en los pacientes con SMD que estaban asociados a un síndrome vasculítico sistémico agudo y/o presentaban crioglobulinas séricas.1,3 Un estudio reciente, con diseño retrospectivo pero gran cantidad de pacientes, no encontró diferencias en términos de supervivencia entre los pacientes con y sin MA, a excepción de los cuatro pacientes que presentaron vasculitis sistémica y/o crioglobulinas.2 Todos ellos, al igual que nuestra paciente, presentaron además evolución de SMD a leucemia mieloide aguda en un intervalo promedio de 6 meses.
1. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127:2391-2405, 2016. [ Links ]
2. de Hollanda A, Beucher A, Henrion D, Ghali A, Lavigne C, Lévesque H, et al. Systemic and immune manifestations in myelodysplasia: a multicenter retrospective study. Arthritis Care Res (Hoboken) 2011; 63:1188-94. [ Links ]
3. Mekinian A, Grignano E, Braun T, Decaux O, Liozon E, Costedoat-Chalumeau N, et al. Systemic inflammatory and autoimmune manifestations associated with myelodysplastic syndromes and chronic myelomonocytic leukaemia: A French multicentre retrospective study. Rheumatology (Oxford). 2016; 55, 291-300. [ Links ]
4. Komrokji RS, Kulasekararaj A, Al Ali NH, Kordasti S, Bart-Smith E, Craig BM, et al. Autoimmune diseases and myelodysplastic syndromes. Am J Hematol. 2016 May;91(5):E280-3. [ Links ]
5. Lambert C, Wu Y, Aanei C. Bone Marrow Immunity and Myelodysplasia. Front Oncol. 2016;6: 172. [ Links ]
6. Saif MW, Hopkins JL, Gore SD. Autoimmune Phenomena in Patients with Myelodysplastic Syndromes and Chronic Myelomonocytic Leukemia. Leuk Lymphoma. 2002 Nov;43(11):2083-92. [ Links ]
7. Giannouli S, Voulgarelis M, Zintzaras E, Tzioufas AG, Moutsopoulos HM. Autoimmune phenomena in myelodysplastic syndromes: a 4-yr prospective study. Rheumatology (Oxford) 2004; 43:626-32. [ Links ]
8. Kennedy JA, Ebert BL. Clinical Implications of Genetic Mutations in Myelodysplastic Syndrome. Journal of Clinical Oncology 2017, 35: 9, 968-974. [ Links ]
9. Mathian A, Miyara M, Cohen-Aubart F, Haroche J, Hie M, Pha M, et al. Relapsing polychondritis: A 2016 update on clinical features, diagnostic tools, treatment and biological drug use. Best Pract Res Clin Rheumatol. 2016 Apr;30(2):316-333. [ Links ]
10. Micheva I, Thanopoulou E, Michalopoulou S, Karakantza M, Kouraklis-Symeonidis A, Mouzaki A, et al. Defective tumor necrosis factor alpha-induced maturation of monocyte-derived dendritic cells in patients with myelodysplastic syndromes. Clin Immunol 2004; 113(December (3)):310-7. [ Links ]
11. Kemta Lekpa F, Kraus VB, Chevalier X. Biologics in Relapsing Polychondritis: A Literature Review. Semin Arthritis Rheum. 2012 Apr;41(5):712-9. [ Links ]
12. Leroux G, Costedoat-Chalumeau N, Brihaye B, Cohen-Bittan J, Amoura Z, Haroche J, et al. Treatment of relapsing polychondritis with rituximab: a retrospective study of nine patients. Arthritis Rheum 2009;61:577-82. [ Links ]

