










Servicios Personalizados
Revista

Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista argentina de reumatología
versión impresa ISSN 0327-4411versión On-line ISSN 2362-3675
Rev. argent. reumatolg. vol.31 no.3 Buenos Aires set. 2020
CASOS CLÍNICOS
Púrpura retiforme, un desafío diagnóstico-terapéutico. Casos clínicos y revisión de la literatura
Retiform purpura, a diagnosic therapeutic challenge. Case reports and literature review
María Noelia Bersano1, Ariana Ringer2, Álvaro Sanabria1, Marcelo Abdala2, Mónica Sacnún1
1Servicio de Reumatología. Hospital Provincial de Rosario. Rosario (Argentina)
2Servicio de Reumatología. Hospital Provincial del Centenario. Universidad Nacional de Rosario. Rosario (Argentina)
Mail de contacto: noebersano@outlook.com / Aruris15151@gmail.com
Resumen
Las manifestaciones cutáneas en las enfermedades autoinmunes son frecuentes y heterogéneas. En algunas de ellas, como en el caso del lupus eritematoso sistémico, la dermatomiositis, la esclerosis sistémica y el síndrome antifosfolípidico son de tal importancia que se incluyen como criterios clasificatorios de la enfermedad. Los diagnósticos diferenciales varían en gravedad, pudiendo en ocasiones presentar riesgo vital, por lo cual se jerarquizan el diagnóstico y tratamiento oportunos. Se describe el caso de una paciente de 22 años con diagnóstico previo de lupus eritematoso sistémico y síndrome antifosfolípídico, que concurre a la consulta con cuadro agudo caracterizado por lesiones cutáneas dolorosas de aspecto necrótico acompañadas de fiebre y livedo reticularis.
Palabras clave: Púrpura Retiforme; Lupus Eritematoso Sistémico; Síndrome Antifosfolípidos
Abstract
Cutaneous involvement is frequent and heterogeneous in autoimmune diseases. In some of them, such as in systemic lupus erythematosus, dermatomyositis, systemic sclerosis and antiphospholipid syndrome, some manifestations are so relevant that are included in the classification criteria. Differential diagnosis ranges in severity. Since the disease may be life-threatening, a prompt diagnosis and treatment are mandatory. We describe a clinical case of a twenty-two-year-old woman with diagnosis of systemic lupus erythematosus and antiphospholipidic syndrome, presenting with acute, painful cutaneous lesions with necrotic aspect, fever and livedo reticularis.
Key words: Retiform Purpura; Systemic Lupus Erytematosus; Antiphospholipidic Syndrome
Introducción
Los pacientes con enfermedades autoinmunes presentan con frecuencia compromiso cutáneo, siendo éste una de las manifestaciones más variadas y desafiantes en cuanto a diagnóstico y tratamientos certeros.
Con respecto al lupus eritematoso sistémico (LES), el 80% de los pacientes presentan lesiones cutáneas durante el curso de la enfermedad1. En este caso suelen clasificarse en específicas y no específicas de lupus. Las lesiones específicas son tan frecuentes e importantes que se hallan presentes en los criterios de clasificación de la enfermedad. En la literatura existen diversas clasificaciones del espectro cutáneo del LES. Lipsker en 2010 presenta una clasificación de lesiones cutáneas del LES en el que diferencia las manifestaciones cutáneas propias de LES de las lesiones con signos de vasculopatía trombótica2. Dentro de este subgrupo se encuentran tromboflebitis, livedo, y necrosis cutánea, entre otras.
En el caso del síndrome antifosfolípido (SAF), éste se caracteriza por trombosis vascular, arterial o venosa; y morbilidad obstétrica en presencia de anticuerpos antifosfolipídicos plasmáticos3. Otras manifestaciones cutáneas sin embargo, no están incluidas en los criterios de clasificación la enfermedad, presentando gran heterogeneidad y suponiendo un desafío diagnóstico y terapéutico para el reumatólogo.
Se presenta una paciente con lesiones cutáneas y fiebre en el contexto de lupus eritematoso sistémico y síndrome antifosfolipídico, planteando diagnósticos diferenciales y tratamientos indicados.
Caso Clínico
Mujer de 22 años de edad con lupus eritematoso sistémico diagnosticado a los 15 años y controlado clínicamente hasta su internación. Los criterios al inicio de su enfermedad fueron rash malar, fotosensibilidad, poliartritis y FAN positivo mayor a 1/160.
Antecedentes personales: colecistectomía y apendicectomía. No se constató etilismo, tabaquismo ni otros hábitos tóxicos. Presentó historia de un aborto espontáneo, y una cesárea por preeclampsia, con recién nacido prematuro y bajo peso para su edad gestacional. Usuaria de dispositivo intrauterino como método anticonceptivo. Al momento de la consulta se encontraba medicada con hidroxicloroquina 400mg (HCQ) diarios y meprednisona 8mg diarios.
Laboratorio del año previo a la consulta: FAN (Hep-2) positivo 1/640 moteado fino; Anti DNA negativo; C3 110 mg/dl (VN: 100 a 180); C4 11 mg/dl (VN: 16 a 40). Proteinuria en orina de 24 horas 0,52 g/24horas. Diuresis 1800 ml.
La paciente fue internada en centro de alta complejidad por cuadro de 7 días de evolución caracterizado inicialmente por astenia, sensación subjetiva de fiebre y dolor en ambos miembros inferiores, agregando en las 24 horas previas, lesiones cutáneas violáceas y dolorosas en muslo izquierdo acompañadas de livedo reticularis en ambos miembros inferiores (Figura 1), con SELENA/SLEDAI 5.
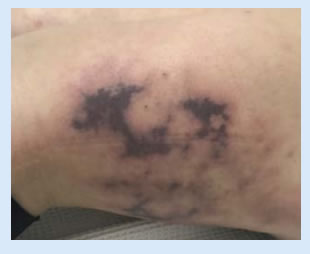
Se realizaron los siguientes estudios:
Laboratorio al ingreso: GB 4,7 x mm3, HB 10,5% (VN: 12 a 15 %) ; HTO 32,1 (VN: 38 a 54%); Plaquetas 143 x; ESD 105 mm; Creatinina 0,84 mg/dl; Uremia 25 mg/dl; Ionograma sérico: Na 140/K 4 meq/l; TGO 16UI/L; TGP 24UI/L; CPK 35; TP 15; KPTT 33; PCR 48 mg/dl; Procalcitonina 0,11 ng/ml; Ferritina 163 ng/ml (VN: hasta 150). Proteinograma con leve hipoalbuminemia y con aumento policlonal de gammaglobulinas.
Serologías: HIV negativo, HbsAg negativo, HCV negativo.
Laboratorio inmunológico: C3 81 mg/dl; C4 14 mg/dl; FAN (+) 1/5120 moteado homogéneo (hígado de rata, confirmado x Hep2); ANTI DNA (-); ENA (+) Sm (+). Por otro lado, AntiS-cl70, Anti centrómero, ANCA C y P, AntiB2 gp: todos negativos; anticardiolipinas: IgM (+) en niveles intermedios, IgG negativo. Anticoagulante lúpico positivo.
Ecocardiograma: FEY 71%, derrame pericárdico leve. Sin otras alteraciones.
Durante la internación se constataron registros febriles de 38°C durante las primeras 48 hs.
Se realizó biopsia de la lesión cutánea y comenzó tratamiento con prednisona 20 mg diarios y anticoagulación con heparina de bajo peso molecular y acenocumarol.
Biopsia cutánea: Se detectan en dermis superficial y en menor grado profunda, vasos capilares con trombosis intraluminal con material acidófilo denso. Escasa presencia de elementos leucocitarios mononucleares en pared de los vasos. Epidermis necrosada. Sin signos de neoplasia. Diagnóstico histopatológico: microangiopatía trombótica.
La paciente evolucionó presentando mejoría del estado general, la fiebre y las lesiones cutáneas (Figura 2), otorgándose el alta luego de siete días de internación, medicada con hidroxicloroquina 200mg/día; prednisona 20mg/día, azatioprina 50mg c/12 hs y acenocumarol 1mg/día.
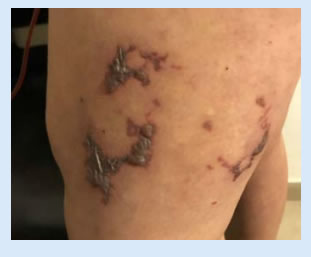
Actualmente continúa con buen estado general, sin rash, alopecia, artritis ni úlceras orales. Las lesiones cutáneas descriptas desaparecieron, a excepción de la livedo reticularis. (Figura 3). Se encuentra medicada con prednisona 20mg/diarios, hidroxicloroquina 400mg/diarios, omeprazol 20mg/diarios, calcio, vitamina D y anticoagulación con acenocumarol. A las 12 semanas de la primera determinación de laboratorio, se solicitaron nuevos anticuerpos antifosfolipidícos, los cuales fueron positivos, confirmando el diagnóstico presuntivo de SAF, asociado a su lupus.
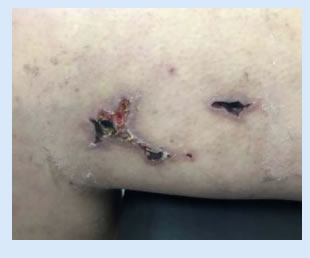
Discusión
Las lesiones purpúricas4 se desarrollan como consecuencia de la oclusión vascular parcial o total con posterior daño vascular. Tanto en la púrpura retiforme, la livedo reticularis y la racemosa, la forma angulada refleja la arquitectura vascular de la piel. La apariencia hemorrágica resulta de la extravasación de glóbulos rojos luego de un período de isquemia. Pueden evolucionar a necrosis cutánea. La livedo reticularis, se debe a una reducción generalizada del flujo sanguíneo, secundario a un proceso patológico sistémico y que origina un patrón reticulado completo, regular, generalmente en miembros inferiores. La livedo racemosa se origina luego de una reducción irregular del flujo sanguíneo, con frecuencia como consecuencia de una obstrucción mecánica, originando un patrón reticulado incompleto con predominio en miembros y tronco. Finalmente, la púrpura retiforme obedece a una obstrucción completa vascular de aspecto serpinginoso o estrellado, con áreas de infarto y necrosis cutánea.
En un paciente con fiebre más púrpura retiforme, se deben considerar múltiples diagnósticos diferenciales4-5, con distintos grados de severidad. Los antecedentes, la clínica acompañante, el laboratorio y otros métodos complementarios ayudarán a guiar el abordaje.
• Patología infecciosa: principalmente meningococos, neumococos, estreptococos y estafilococos aureus, habitualmente en el contexto de infecciones diseminadas, endocarditis y émbolos sépticos.
• Patología cardiológica: émbolos de colesterol y mixoma auricular.
• Patología oncológica: carcinoma renal, el linfoma intravascular y el mieloma múltiple.
• Patología hematológica: policitemia vera, trombocitosis esencial, púrpura trombocitopénica, síndrome de Snnedon, microangiopatía trombótica clásica (MAT), síndrome antifosfolípido catastrófico (CAPS), coagulación intravascular diseminada (CID).
• Patología reumatológica: vasculitis, manifestaciones cutáneas de LES y SAF, principalmente.
La púrpura retiforme habitualmente se observa en vasculitis de vaso mediano6. Si bien la poliarteritis nodosa, se ha descripto con hallazgos de livedo reticularis o racemosa, presenta con mayor frecuencia nódulos, infartos cutáneos y ulceraciones, no descriptas en la paciente, así tampoco otra clínica sugestiva como afectación renal o neuropática. En contraste, las vasculitis de vaso pequeño6 suelen presentarse con púrpura palpable no retiforme, con tendencia a la ulceración. Para el diagnóstico de una vasculitis ANCA limitada a piel, es mandatorio la positividad de anticuerpos ANCA. La crioglobulinemia se manifiesta como isquemia cutánea que rápidamente se ulcera por el depósito de precipitados de crioglobulinas. Otras etiologías a considerar son las vasculitis asociadas a drogas.
Clásicamente, la afectación cutánea de LES7-9 se describe como lesiones específicas y no específicas. Entre estas últimas, se destacan las vasculitis de vaso pequeño y mediano, las lesiones vasculopáticas en relación a anticuerpos antifosfolípidos o crioglobulinas y la vasculopatía livedoide.
Por su parte, en el SAF10-15 pueden observarse lesiones muy variadas, desde petequias, úlceras, ampollas hemorrágicas, livedo reticularis, racemosa, púrpura retiforme, necrosis, gangrena digital, lesiones pseudovasculíticas (nodulares, etc) y hasta vasculopatía livedoide.
La vasculopatía livedoide6-9, también descripta como vasculitis livedoide, vasculitis segmentaria hialina, PURPLE (acrónimo en inglés de “painful purpuric ulcers with reticular patterning on the lower extremities”), o atrofia blanca (en estadios avanzados), es una vasculopatía trombo-oclusiva cutánea de vaso pequeño, dolorosa, tendiente a la cronicidad, como consecuencia de un aumento de la coagulación con disminución de fibrinólisis, pudiendo consumir complemento. Hay afectación de dermis y se manifiesta con mayor frecuencia en miembros inferiores. Clínicamente puede evidenciarse como livedo reticularis, livedo racemosa, púrpura retiforme, ulceración y más tarde, atrofia blanca, de acuerdo al estadio. Puede o no asociarse con factores de riesgo trombótico, como LES y SAF. El diagnóstico se confirma por biopsia. En la anatomía patológica se observan trombos intraluminares de fibrina, proliferación endotelial y degeneración hialina subintimal. Puede existir extravasación de glóbulos rojos. La ausencia de infiltrados inflamatorios perivasculares o leucocitoclastia la diferencian de las vasculitis.
Por su parte, Ashershon14 define el concepto de síndrome antifosfolípido microangiopático (MAPS por sus siglas en inglés) como un amplio abanico de patologías, que incluye a aquellos pacientes con microangiopatía trombótica y anticuerpos antifosfolípidos (ApL) demostrables. Las presentaciones tienen gravedad variable, desde livedo reticularis, púrpura retiforme aislada, trombocitopenia y vasculopatía livedoide, hasta cuadros de mayor severidad, como el síndrome de Sneddon, la púrpura trombocitopénica trombótica, SAF catastrófico, MAT clásica, síndrome HEELP y CID. Se postula que tendrían distintos factores desencadenantes, como infecciones, drogas y la propia actividad de la enfermedad autoinmune, por ejemplo, LES.
En el caso presentado se plantea como posible diagnóstico una vasculopatía livedoide, en contexto de síndrome antifosfolípido microangiopático y lupus eritematoso sistémico activo, como consecuencia de microtrombosis.
El tratamiento realizado consistió en hidroxicloroquina, azatioprina 100 mg/día, prednisona 20 mg/día y anticoagulación con heparina de bajo peso molecular y acenocumarol 1mg/día, con buena evolución. Debido a que la paciente presentaba actividad lúpica moderada, se indicó tratamiento inmunosupresor, pero además se ponderó la necesidad de anticoagulación oportuna en dichos casos, para controlar los eventos trombóticos.
No hay estudios randomizados sobre el tratamiento de la vasculopatía livedoide. La evidencia disponible recomienda que, ante la presencia de patología trombótica subyacente, como en el caso presentado, un síndrome antifosfolípido microangiopático, debe iniciarse anticoagulación de manera precoz. Por el contrario, de no existir patología trombótica, se recomiendan aspirina y pentoxifilina. Debido a que puede existir una interrelación entre la vasculopatía livedoide, el síndrome antifosfolípido microangiopático y la actividad del LES, el tratamiento inmunosupresor también está indicado y la elección del inmunosupresor dependerá de las manifestaciones clínicas. Además, se sugiere manejo del dolor y protección cutánea para prevenir infecciones. Otros tratamientos descriptos en la literatura son corticoides a altas dosis, plasmaféresis, rituximab, inmunoglobulinas endovenosas y eculizumab, entre otros, en casos severos y refractarios y, de acuerdo a la patología autoinmune de base9-14.
Debido a que la vasculopatía livedoide puede ser expresión de múltiples procesos patológicos subyacentes y con distintos grados de severidad, desde lesiones cutáneas banales hasta compromiso trombótico multisistémico, es un desafío al inicio del cuadro predecir su evolución. Los diagnósticos diferenciales son inicialmente clínicos, pudiendo ser corroborados posteriormente con laboratorio, biopsia y otros estudios complementarios. Un alto nivel de sospecha y el tratamiento precoz con inmunosupresión y anticoagulación, son claves en la morbimortalidad de esta entidad. También se destaca la necesidad de un seguimiento cercano luego del evento inicial, por la posibilidad de recaídas u otras complicaciones trombóticas9-14.
Conclusión
Las manifestaciones cutáneas en las enfermedades autoinmunes representan un desafío diagnóstico. En el caso presentado, una lesión de aspecto necrótica en contexto de SAF y LES plantea un abanico de patologías posibles, con distintos grados de severidad, algunas de los cuales implican riesgo de vida de no ser tratadas a tiempo. Es importante para los reumatólogos el conocimiento de estas entidades, ya que su tratamiento consiste en anticoagulación, más allá de la inmunosupresión si existe patología autoinmune acompañante. Se jerarquiza la necesidad de instaurar una terapéutica en forma rápida y eficaz seguida de un control clínico continuo para prevenir la evolución a cuadros más severos y con alta morbimortalidad.
1. Ribero S, Lipsker D, Borradori L. The Spectrum of Cutaneous Manifestations in Systemic Lupus Erythematosus and Novel Classification. Rare Diseases of the Immune System 2016;77-94. [ Links ]
2. Lipsker D. The need to revisit the nosology of cutaneous lupus erythematosus. Lupus. 2010; 19(9):1047-1049. [ Links ]
3. Miyakis S, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4:295-306. [ Links ]
4. Wysong A, Venkatesan F. An approach to the patient with retiform purpura. Dermatol Ther. 2011;24(3):151-72. [ Links ]
5. Carpio-Chaname C, Vilchez-Rivera S, Failoc-Rojas VE. Síndrome de Sneddon asociado a síndrome antifosfolipídico: descripciones clínicas y revisión de la literatura. Revista Colombiana de Reumatología. 2017; 24(3):185-188. [ Links ]
6. Zelger B, Chen K, Requena L, Piette W, Sunderk CH, Carlson JA, et al. Nomenclature of Cutaneous Vasculitis Dermatologic Addendum to the 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheumatol. 2018;70(2):171-84. [ Links ]
7. Kuhn A, Landmann A, Bonsmann G. Cutaneous Lupus Erythematosus. Systemic Lupus Erythematosus: Basic, Applied and Clinical Aspects. Elsevier; 2016. 333-339 p. [ Links ]
8. Chong BF, Werth VP. Skin Disease in Cutaneous Lupus Erythematosus. Ninth Edition. Lupus Erythematosus and Related Syndromes. Elsevier Inc; 395-406 p. [ Links ]
9. Grinnell M, et al. Retiform non-blanchable purpuric plaques in a patient with systemic lupus erythematosus. Lupus. 2019;28:1013-6. [ Links ]
10. Weinstein S, Piette W. Cutaneous Manifestations of Antiphospholipid Antibody Syndrome. Hematology/oncology clinics. 2008; 22:67-77. [ Links ]
11. Kroshinsky D, Stone JH, Bloch DB, Sepehr A. Case 5-2009: A 47-Year-Old Woman with a Rash and Numbness and Pain in the Legs. N Engl J Med. 2009;360:711-20. [ Links ]
12. Fanouriakis A, Kostopoulou M, Alunno A, Aringer M, Bajema I, Boletis JN, et al. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis. 2019;78:736-45. [ Links ]
13. Micieli R, et al. Treatment for Livedoid Vasculopathy: A Systematic Review. JAMA Dermatol 2018;154(2):193-202. [ Links ]
14. Asherson RA. New subsets of the antiphospholipid syndrome in 2006: “ PRE-APS ” (probable APS) and microangiopathic antiphospholipid syndromes (“ MAPS ”). Autoimmun Rev. 2006;6:76-80.
15. Cervera R. Antiphospholipid syndrome. Thrombosis Research. 2017;151(Suppl 1):S43-S47. [ Links ]

