











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome antisintetasa (SAS) es una enfermedad autoinmune poco frecuente, definida como nueva entidad en 1990. Recientemente (2011) se introdujeron criterios diagnósticos que facilitaron su reconocimiento clínico1,2. Se manifiesta a menudo con enfermedad intersticial pulmonar (EPI), compromiso muscular y artritis3-4. El objetivo de esta presentación es la difusión de una entidad poco prevalente y la revisión de la literatura.
Caso clínico
Paciente de sexo femenino, 45 años de edad, presenta cuadro clínico progresivo que inició 3 años atrás caracterizado por mialgias generalizadas, asociado a poliartralgias a nivel de cintura escapular, rodillas y articulaciones interfalángicas proximales. Refiere fenómeno de Raynaud en dedos de ambas manos y edema bilateral en manos y pies con mejoría espontánea. Al inicio del cuadro, consulta a otro centro donde se identifica, como hallazgo positivo en laboratorio, elevación de transaminasas y anticuerpos antinucleares (ANA) 1/320, anti-La y anti-Ro positivos. Abandona el seguimiento y suma síntomas de forma insidiosa: astenia, adinamia, hiporexia, disminución de fuerza muscular proximal de miembros superiores e inferiores hasta impedir actividades de la vida cotidiana, xerostomía e hipertrofia parotídea bilateral con dificultad para la apertura bucal completa. En el último año comienza con pérdida de peso de aproximadamente 5 kg y episodios de disnea capacidad funcional II (CF II).
Se le indica tratamiento con corticoides (prednisona 25 mg/día) y por persistencia del cuadro consulta a nuestro centro.
Al examen físico presenta debilidad muscular de miembros superiores e inferiores 3/5 proximal y 5/5 distal, signo de Gottron a nivel de articulaciones metacarpofalángicas (Figura 1A), rash fotosensible a nivel facial (Figura 1C), y eritema en V en cuello, signo de Shawl, eritema periungueal y lesión nodular en pierna izquierda (Figura 1B).
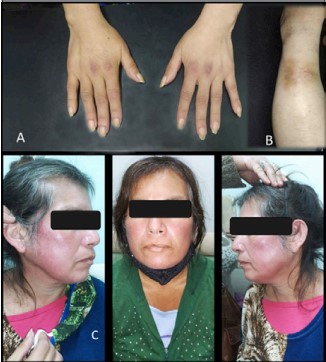
Figura 1: Se observa: A) signo de Gottron en resolución a nivel de articulaciones metacarpofalángicas; B) lesión nodular en pierna izquierda; C) rash fotosensible a nivel facial
En el laboratorio se evidencia: elevación de transaminasas x 3; creatina kinasa (CK) elevada: 5.326 UI/L (normal hasta 170 UI/L); eritrosedimentación: 64 mm/H (normal hasta 30 mm/H); proteína C reactiva: 3,8 mg/dL (normal hasta 0,5); serologías para sífilis, Chagas, hepatitis B, C y virus de inmunodeficiencia humana (VIH) negativas. En el proteinograma electroforético se detecta hipergammaglobulinemia policlonal.
Laboratorio inmunológico: factor reumatoideo: (+) 51,7 UI/ml; ANA: 1/640 con patrón moteado; antígenos extraíbles del núcleo (ENAs): antiRo-52 (+); anti-Ro-60 (+), anti-La (+) y anti-Jo1 (+). Antipéptido citrulinado (aCCP), anti-DNA, antiSM, anti-RNP, anti Scl-70, anticentrómero, antiribosomal P y antihistonas, todos negativos.
Se interpreta el cuadro como SAS con fenotipo de dermatomiositis asociado a síndrome de Sjögren. Este último diagnóstico se objetiva luego de una prueba positiva de sialometría.
Se complementa el estudio con tomografía axial computada (TAC) de tórax de alta resolución, con evidencia de áreas de engrosamientos septales a nivel periférico a predominio de ambos lóbulos inferiores como signos de intersticiopatía (Figura 2A). Se realiza resonancia magnética nuclear (RMN) de cintura escapular y pelviana que informa edema intersticial a nivel del glúteo mayor y medio del lado derecho (Figura 2B).

Figura 2: Se observa: A) en TAC de tórax, áreas de engrosamientos septales a nivel periférico con predominio de ambos lóbulos inferiores; B) en RMN de cintura escapular y pelviana, edema intersticial a nivel de glúteo mayor y medio del lado derecho
Se aumenta la dosis de glucocorticoides a 50 mg/día ajustada al peso del paciente y, debido al compromiso pulmonar, se coadyuva con metotrexato 15 mg semanal/VO (vía oral).
Evoluciona favorablemente con mejoría de debilidad muscular y continúa seguimiento ambulatorio por Reumatología.
Discusión
En 1990, Margueri et al. definieron el SAS como una nueva entidad. A su vez Dalakas y Holhfeld propusieron criterios diagnósticos que se revisaron en 2003. No obstante, no fue hasta 2010 cuando Connors et al. y Solomon en 2011 introdujeron criterios diagnósticos: presencia de un anticuerpo antisintetasa (aAS), sumados a dos criterios mayores (evidencia de miopatía inflamatoria según los criterios de Bohan y Peter), EPI no atribuida a otras causas o con un criterio mayor y dos menores (artritis, fenómeno de Raynaud y manos de mecánico)1,2,4,5. Es importante recalcar que la ausencia aAS no excluye el SAS dado que los niveles de autoanticuerpos fluctúan según la actividad de la enfermedad2,6.
La prevalencia global de las miopatías inflamatorias idiopáticas (MII) se encuentra, según las distintas series, entre 2,4 a 33,8 casos/100.000 habitantes, y la incidencia oscila entre 1,16 a 19 casos nuevos/millón de habitantes/año7-8.
El Registro Europeo de Miopatías Inflamatorias (EuroMyositis Registry), con 3.067 pacientes, reportó una prevalencia de 17% para SAS, siendo el tercer subtipo más frecuente después de la dermatomiositis (31%) y la polimiositis (27%), predominando en caucásicos9,10,11. La prevalencia reportada para SAS en otras poblaciones fue similar: de 19,4% en una cohorte China y de 14,1% en una cohorte estudiada en EE.UU.10,11.
El SAS afecta con mayor frecuencia el sexo femenino, con una relación mujer-varón 2:14-12. Generalmente se presenta durante la cuarta década de la vida, al igual que nuestro caso clínico.
Su patogenia no se conoce con certeza. Se han propuesto varias hipótesis y la más aceptada es la pérdida de la tolerancia inmunitaria celular debido a los múltiples anticuerpos séricos circulantes que causan injuria muscular con el consiguiente desarrollo de autoanticuerpos13-14. Hasta el momento se describieron ocho aAS dirigidos contra la ARNt sintetasa, enzima citoplasmática que aporta aminoácidos al ribosoma durante la traslación de polipéptidos. El más común es el anti-histidil-ARNt-sintetasa o anti-Jo1, que se encuentra presente en el 60,3% de los pacientes2,3.
Coincidente con las manifestaciones clínicas de nuestra paciente, la tríada clásica de síntomas descritos en el SAS incluye: EPI, miositis y artritis. Sin embargo, solo una minoría de los pacientes (19% en un estudio) exhibe la tríada completa al inicio de la enfermedad2,12,15.
La EPI es una de las características más frecuentes del SAS y tiene una prevalencia del 68-94,4% según diversas series10,11,16,17. Las manifestaciones respiratorias pueden variar desde formas leves (tos seca persistente) hasta formas complicadas (insuficiencia respiratoria). La expresión tomográfica de alta resolución en la EPI se manifiesta habitualmente como: neumonía intersticial organizada/criptogénica (NOC) que, por lo general, tiene buena respuesta al tratamiento con esteroides, o neumonía intersticial no específica (NINE), que precisa de tratamiento con esteroides e inmunosupresores, y su respuesta depende del grado o estadio de fibrosis3,4.
La tomografía computarizada de alta resolución es fundamental para el diagnóstico y seguimiento de los pacientes con SAS dado que la presencia de EPI es predictora de mal pronóstico2,3,4. Se describió que los aAS (anti-PL7 y anti-PL12 así como anti-Ro52) se relacionan con empeoramiento de la función pulmonar y mala respuesta al tratamiento1,2,3,4. La copositividad anti-Jo1 y anti-Ro52 se asocia con una afectación pulmonar, muscular y articular más grave de inicio temprano, tal como se evidencia en nuestra paciente2,10,11.
El autoanticuerpo anti-Ro52 en pacientes con anti-Jo1 alcanzó el 65% en un informe publicado10,11.
Es importante que ante un diagnóstico de SAS se evalúe la concomitancia con anti-Ro52 porque puede ayudar a estratificar a aquellos pacientes que tienen más probabilidades de desarrollar una enfermedad grave10.
En cuanto al compromiso muscular, si bien se manifiesta en la mayoría de los casos, su inicio puede ser de forma completa o incompleta. El subconjunto de pacientes que presenta SAS incompleto simula carecer de compromiso muscular, sin embargo pueden evidenciarse niveles elevados de enzimas musculares sin déficit de fuerza o limitación funcional, denominados “hipomiopáticos”. Este hallazgo se observa en aproximadamente el 25% de los pacientes con SAS2,11,16,18.
Por su parte, los pacientes con SAS completo muestran compromiso muscular que clínicamente se manifiesta por la presencia de debilidad muscular progresiva proximal de cinturas escapular y pelviana. La actividad se evalúa mediante elevación de las enzimas musculares (CK y la aldolasa). La RMN de cintura escapular y pelviana es útil para identificar edema muscular que podría ser compatible con inflamación muscular2,3,4.
Las manifestaciones articulares se expresan en forma de poliartritis bilateral, simétrica no erosiva en el 58-70% de los casos2,16.
Si bien las lesiones cutáneas características del SAS son las “manos de mecánico” (lesiones agrietadas, descamativas en el borde radial de los dedos y en la palma de las manos), estudios actuales encontraron que la misma está presente en el 16-21% de los casos. Además, se ha descrito recientemente otra manifestación cutánea conocida como “pies de excursionista” de iguales características a las de las manos, pero en los pies2.
Fenotípicamente el SAS puede presentar lesiones características de dermatomiositis: pápulas de Gottron, signo de Gottron, eritema en Heliotropo, signo del Shawl, eritema facial que compromete surco nasogeniano, como fue el caso de nuestra paciente quien presentó varias de estas lesiones cutáneas características de la dermatomiositis.
El fenómeno de Raynaud afecta a más de la mitad de los pacientes, al igual que en otras patologías del tejido conectivo2,4.
El tratamiento consiste en la administración de corticoides en dosis de meprednisona/prednisona 1 mg/Kg/día VO, asociado a metotrexato semanal u otros inmunosupresores, dependiendo si predomina el compromiso muscular o respiratorio (azatioprina, micofenolato de mofetilo, ciclofosfamida, ciclosporina A o rituximab), y según a la respuesta clínica del paciente1,2,3,4.

