Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
La esclerosis sistémica progresiva (ESP) es una enfermedad autoinmune, sistémica y es el fenómeno de Raynaud la característica clínica más frecuente, además de la presencia de esclerodactilia, ulceraciones digitales distales y el habitual compromiso digestivo y pulmonar1. Según Le Roy, la ESP se clasifica en tres subtipos: variedad cutánea limitada, cutáneo difusa y esclerodermia sin esclerodermia.
Por otro lado, los síndromes esclerodermiformes (SE) son un grupo de entidades que producen cambios en la piel similares o parecidos a los de la esclerodermia, pero con mecanismos etiopatogénicos totalmente distintos. Son numerosas las enfermedades que pueden generar los SE: hipotiroidismo, diabetes mellitus, porfiria cutánea tarda, fenilcetonuria, síndrome carcinoide, toxicidad farmacológica, escleredema de Buschke, enfermedad injerto contra huésped, entre otras2,3.
Las porfirias son un grupo de enfermedades heterogéneas causadas por una deficiencia genética o adquirida de enzimas que regulan la síntesis del hemo. La presencia o ausencia de fotosensibilidad (cutánea) diferencia a las porfirias en cutáneas y no cutáneas. Existen cinco tipos de porfirias cutáneas: porfiria cutánea tarda (PCT), porfiria variegata, coproporfiria hereditaria, protoporfiria eritropoyética y porfiria eritropoyética congénita. De todas, la PCT es, por lejos, la porfiria cutánea más frecuente con una incidencia de 1 en 10000 personas. Su cuadro clínico incluye fotosensibilidad, ampollas cutáneas, hipertricosis e hiperpigmentación. La afectación hepática crónica suele ser leve o inexistente. Su diagnóstico se hace mediante detección de porfirinas en orina, sangre y heces. El tratamiento de una PCT se basa en sangrías y dosis medias a bajas de hidroxocloroquina4. Los diagnósticos diferenciales en medicina son un punto central que debe tenerse en cuenta en todo momento, principalmente en pacientes con enfermedades raras o poco prevalentes. Es deber del reumatólogo saber distinguir acertadamente una esclerodermia de un SE.
A continuación, se presenta el caso de un varón con esclerodactilia y lesiones cutáneas cicatrizales que simularon a la perfección el cuadro clínico de una ESP.
Caso clínico
Varón de 48 años, natural de Cajamarca, Perú, agricultor, grado de instrucción primaria completa, no refiere enfermedades crónicas, niega hábitos nocivos ni reacciones adversas medicamentosas.
Durante su infancia inicia formación de ampollas cutáneas, principalmente a la exposición solar, a predominio distal, con cicatriz residual, agregándose fragilidad cutánea asociada a pigmentación en zonas fotoexpuestas. Dichas alteraciones las mantiene hasta la actualidad, con predominio del cuadro en las orejas y en el dorso de las manos.
Durante la adolescencia inicia resorción lenta y progresiva pérdida de falanges distales de todos los dedos de las manos, asociado a esclerosis cutánea en cara y extremidades; además refiere hipertricosis en zona preauricular y en antebrazos, con artralgias sin artritis/sinovitis. El paciente niega fenómeno de Raynaud y refiere dificultad para ganar peso, sin embargo, niega cansancio o malestar general. Al iniciar su segunda década de vida observa que su orina y heces se tornan oscuras con la luz solar. Percibe, además, que al tomar suplementos de hierro e ingesta de alcohol se exacerba el cuadro de fotosensibilidad.
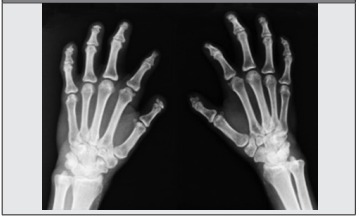
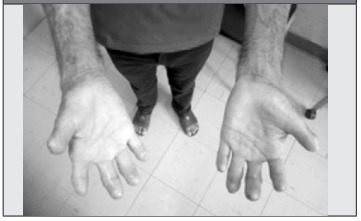
Por todo el cuadro anterior acude al consultorio de Reumatología, donde se evidencia esclerodactilia con cicatrices visibles asociada a falta parcial de falanges distales en todos los dedos, con pérdida de tejido ungueal (Figura 1 y Figura 2), así como esclerosis cutánea facial muy llamativa (Figura 3), por lo cual se propone en primera instancia el diagnóstico de ESP y se solicita analítica básica que arroja disfunción hepática con elevación de transaminasas x 3 aproximadamente (AST: 125 u/ml, ALT: 158 u/ ml, GGTP: 108 mg/dl), siendo el resto de los parámetros de laboratorios y radiografía de tórax normales.

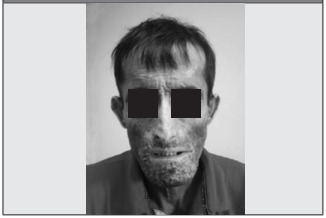
Se solicitan anticuerpos antinucleares (ANA), antitopoisomerasa (SCL-70) y anticentrómero, que resultan todos negativos. Se reinterpreta el caso y se sospecha porfiria. Se piden marcadores para porfiria en orina de 24 h, con resultados positivos: uroporfirina: 86 ug (VN: <2 ug), protoporfirina: 127 ug (VN: 16.60 ug) y valores elevados de ferritina: 1.600 ng/ml (VN: 10-300 ng/ml). Además, se observa oscurecimiento de orina a la luz solar. Debido a la facies leonina, se solicita serología para lepra, con resultado negativo, la serología para virus de hepatitis B y C también resultan negativos.
Por la ausencia de antecedentes familiares, de cuadro neurológico, y de compromiso hepático y de piel, se hace diagnóstico de PCT y se inicia tratamiento mediante flebotomías e hidroxocloroquina 200 mg semanal. Actualmente el paciente mantiene el tratamiento indicado, con mejoría ostensible de fotosensibilidad y disminución de las lesiones ampollares, además se observa disminución progresiva de los valores de transaminasas.
Discusión
La presente publicación es el primer reporte en Perú sobre un paciente con PCT que simuló una ESP. Destacamos la importancia de reconocer los SE y descartar los diversos diagnósticos diferenciales ante un paciente que se presenta con esclerodermia, ANA negativo, anticuerpos específicos negativos y niegue fenómeno de Raynaud.
La PCT es la única porfiria que puede presentarse en ausencia de una mutación netamente hereditaria, por ende, es un trastorno adquirido donde interactúan factores conductuales, genéticos, ambientales e infecciosos para causar una deficiencia de la enzima hepática uroporfirinógeno descarboxilasa (UROD)5.
Aunque la PCT se considera dentro de los SE, la presencia de esclerosis cutánea no es común en estos pacientes. Khayat et al publicaron un estudio de 6 pacientes mujeres con PCT, todas con piel indurada, en quienes las biopsias demostraron morfea, pero los niveles elevados de porfirinas en orina orientaron el diagnóstico seguro, si bien tardío, hacia PCT6.
Otros autores, Tkachenko et al, reportaron dos casos de PCT con lesiones cutáneas induradas y recalcaron la importancia de una observación clínica minuciosa y la sospecha clínica. Es interesante informar que uno de los pacientes tuvo una ANA 1/1280, sin embargo valores elevados de uroporfirina en orina y el cuadro clínico característico hicieron el diagnóstico de PCT7.
Wallaey et al destacaron que la presencia de una pseudoesclerodermia en la PCT es excepcional, pudiendo ser una complicación grave o una forma inicial de presentación en estos pacientes. Además, mencionaron que al descender niveles de porfirina debido a un correcto tratamiento, también mejora la esclerosis cutánea existente8.
Conclusiones
Se deberá considerar a PCT como un probable diagnóstico ante un paciente con fotosensibilidad, lesiones ampollares y esclerosis cutánea asociado a acroosteólisis de los dedos de las manos (Figura 4), debiendo solicitar uroporfirinas en orina de 24 h para confirmar la sospecha.