











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
El compromiso de sistema nervioso central (SNC) en el síndrome de Sjögren primario (SSp) es infrecuente. La prevalencia reportada es de aproximadamente 10-20% y se presenta más frecuentemente como encefalopatía o meningitis aséptica1-3.
Zhao et al reportaron, en una cohorte retrospectiva de pacientes con encefalitis autoinmune (EA) con anticuerpos positivos, que el 8% tenía EA concomitante, dentro de los cuales el 0,39% tenía SSp2. En esta cohorte, el anticuerpo contra la proteína 1 inactivada del glioma rica en leucina o LGI1 (proteína inactivada del glioma rica en leucina 1, del complejo VGKC) fue el más frecuentemente asociado, pero en otro estudio multicéntrico fue más frecuente el anticuerpo anti N-metil-D-aspartato (NMDA-R)4. Es posible que las diferencias en la frecuencia de los anticuerpos se deban a la disponibilidad y el método diagnóstico utilizado (kit comercial o prueba casera)4.
La EA se caracteriza clínicamente por cambios en el comportamiento, psicosis, convulsiones, déficit de memoria, trastornos cognitivos, movimientos anormales, disautonomía y disminución del nivel de consciencia. Las manifestaciones disautonómicas se asocian con los anticuerpos NMDA-R y la proteína símil 2 asociada a contactina del complejo VGKC o CASPR2. Los síntomas se presentan generalmente de forma subaguda, en días a semanas4,7-9. La resonancia magnética nuclear (RMN) de cerebro es anormal en el 30% de los afectados, mostrando principalmente compromiso en las regiones cortical, subcortical o cerebelosa en secuencias FLAIR5.
Caso clínico
Mujer de 55 años con diagnóstico de SSp que presentó en su evolución ojo seco, boca seca y poliartritis no erosiva desde 2016. En el laboratorio se detectó ANA título 1/320 con patrón nuclear moteado, anti-Ro y anti-La positivos, anti-RNP positivo a bajos títulos, anticitrulina positiva e hipocomplementemia crónica.
Antecedentes clínicos de relevancia: fibromialgia, lumbalgia crónica, esteatosis hepática, enfisema centrolobulillar y cefalea crónica. Su medicación habitual era hidroxicloroquina 200 mg/día por medio, pregabalina 150 mg cada 12 h, alprazolam 2 mg/noche y pilocarpina cada 8 h.
La paciente consultó con el Departamento de Emergencias en mayo de 2022 por cefalea difusa y episodios de confusión de 4 días de evolución, con sensación de molestia en el cuero cabelludo y fotofobia. Al interrogatorio dirigido, refería episodios de cacosmia, alucinaciones visuales tipo zoopsia y dificultades en la memoria de corto plazo.
Al ingreso estaba febril e hipotensa (TA 80/40 mm Hg), somnolienta y con bradilalia, síntomas que mejoraron luego de la infusión de cristaloides. Durante la internación tuvo alucinaciones visuales y olvidos frecuentes.
Se realizaron las siguientes determinaciones: PCR SARS-CoV-2, urocultivo y hemocultivos que resultaron negativos. También se efectuó una punción lumbar que informó líquido cefalorraquídeo (LCR) con proteínas 0,5 g/l, leucocitos 78/campo (95% PMN), hematíes 0/campo, glucorraquia 64 mg/dl (glucemia 106 mg/dl). El cultivo de LCR fue negativo para gérmenes comunes, hongos y micobacterias. Los hallazgos del laboratorio se detallan en la Tabla 1.
La tomografía computada de encéfalo fue normal. Se realizó RMN de encéfalo sin contraste que evidenció múltiples imágenes de comportamiento restrictivo, de señal hiperintensa en T2 y FLAIR, a predominio córtico-subcortical a nivel occipital bilateral, hemisferio cerebeloso derecho y parietal derecho (Figura 1). Se realizó angiorresonancia cerebral que excluyó compromiso de vasos intracraneanos.
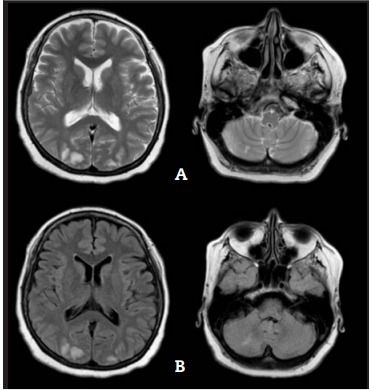
Figura 1: Resonancia magnética nuclear cerebral sin contraste. Se observan múltiples imágenes de comportamiento restrictivo, de señal hiperintensa en T2 (A) y FLAIR (B), a predominio córtico-subcortical a nivel occipital bilateral, hemisferio cerebeloso derecho y parietal derecho.
Se descartó infección por enterovirus, varicela, herpes, virus JC mediante PCR en LCR. Por no poder descartar infección por listeria, recibió tratamiento empírico con ampicilina.
Se realizaron los siguientes autoanticuerpos: anti-NMDA-R, anti-AMPA-R1/R2 (receptor de glutamato tipo AMPA, subunidad 1/2), anti-CASPR2, anti-LGI1, anti-GABA B1/B2 (receptor de ácido gamma amino butírico tipo B, subunidades 1 y 2) y anti-DPPX (dipeptidil aminopeptidasa like proteina-6). Todos resultaron negativos en sangre y LCR según correspondiera. También fueron negativos anti-DPPX, aquaporina-4 y anti-MOG (mielina oligodendrocito glicoproteína) en LCR. Se obtuvo IgG total en LCR de 0,7 mg/dl (VN 1-3 mg/dl). No se observó presencia de bandas oligoclonales en el LCR ni en sangre.
Se interpretó el cuadro como meningoencefalitis aséptica de probable origen autoinmune, seronegativa. Se administraron 5 pulsos de 1 g de metilprednisolona. La paciente se externó con meprednisona 1 mg/kg/día que se descendió progresivamente hasta 4 mg/día en el transcurso de 12 meses.
A los 3 meses del diagnóstico, se realizó RMN de encéfalo de control sin contraste que evidenció gliosis de pequeño tamaño occipital como único signo secuelar (Figura 2). Presentó mejoría de las alteraciones cognitivas de forma progresiva, pero con persistencia de cefaleas leves ocasionales y episodios de anomia en el control anual.
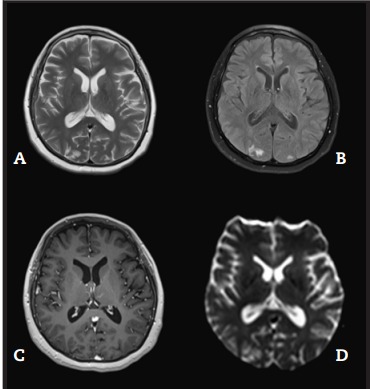
Figura 2: Resonancia magnética nuclear cerebral (A) T2, (B) T2 fluid Dark, (C) T1 con gadolinio, (D) secuencia de difusión 3 meses después de iniciar tratamiento. Se observa pequeña área focal de encefalomalacia en precuña parietal derecha. Foco de gliosis a nivel subcortical en precuña parietal izquierda y cerebelo derecho.
Discusión
El compromiso del SNC en SSp ocurre en menos del 20% de los pacientes1-3. La EA, por otro lado, es un trastorno neurológico con una amplia variedad de síntomas que pueden ser discapacitantes con pronóstico variable. La EA seronegativa es probablemente causada por un autoanticuerpo desconocido o una enfermedad mediada por células T, por lo tanto, representa un grupo potencialmente heterogéneo de trastornos6.
Existen pocos casos en la literatura que describan la asociación entre el SSp y la EA seronegativa7. Esta puede presentarse concomitantemente con el inicio de la enfermedad y el diagnóstico se basa en tres criterios: 1) el inicio subagudo (menos de 3 meses de déficit de memoria de trabajo, estado mental alterado o síntomas psiquiátricos; 2) al menos uno de los siguientes: alteraciones focales en el SNC, convulsiones no explicadas, pleocitosis en LCR (>5 leucocitos/mm3), alteraciones en la RMN sugestivas de encefalitis; 3) exclusión de causas alternativas7,8. Aunque se han descrito 10 anticuerpos asociados9,6, se estima que un 40% de los pacientes puede tener anticuerpos negativos4.
Los síntomas más frecuentemente reportados son: alteraciones mnésicas (93,2%), convulsiones (81%), alteraciones de la conciencia (80,3%), inestabilidad/ataxia (79,6%), síntomas psiquiátricos (76,2%) y alteraciones del lenguaje (71,4%), con leves variaciones entre los subtipos10. Es frecuente el hallazgo de anormalidades en el LCR (90,55%), principalmente pleocitosis (57%). El 91% tiene anormalidades en la RMN cerebral, que incluyen lesiones de atenuación en T2, con restricción de la difusión, principalmente en la corteza (80,3%) y sustancia blanca (42,9%), además del refuerzo con gadolinio10.
A diferencia de la EA seropositiva, en la EA seronegativa el inicio suele ser más rápido (en pocas semanas), y los pacientes presentan marcada disfunción cognitiva con compromiso de la memoria de corto plazo y desorientación11.
Nuestra paciente manifestó principalmente alteraciones cognitivas (confusión, anomia, memoria de corto plazo), cefalea (con fotofobia, algias en cuero cabelludo), cacosmia y zoopsias. Todos estos síntomas son frecuentemente reportados por diferentes autores y son parte de los criterios diagnósticos, a excepción de la cefalea8,10. Nuestra paciente no presentó actividad comicial, un síntoma frecuentemente encontrado en esta enfermedad. En la RMN de encéfalo se evidenció señal hiperintensa en T2 y FLAIR, a predominio córtico-subcortical a nivel occipital bilateral, hemisferio cerebeloso derecho y parietal derecho, hallazgos compatibles con encefalomielitis autoinmune seronegativa5,10.
En relación con el tratamiento, no existen hasta el momento ensayos clínicos aleatorizados que comparen distintas alternativas terapéuticas. Sin embargo, las recomendaciones actuales enfatizan no retrasar el tratamiento cuando existe la sospecha diagnóstica y se ha descartado la etiología infecciosa6. Un estudio retrospectivo, realizado en el Hospital Universitario Nacional de Seúl que incluyó 147 pacientes con EA seronegativa, concluyó que el retraso mayor a un mes en la instauración del tratamiento se asocia a un peor curso clínico a los 2 años (definido por escala de Rankin modificada)10. La selección del inmunosupresor debe basarse en la evidencia disponible, la presentación sindrómica específica y las comorbilidades.
El tratamiento empírico con metilprednisolona endovenosa a dosis de 1 g/día por 3-7 días (solo o en combinación con otro agente inmunosupresor) es un abordaje adecuado para lograr un efecto antiinflamatorio e inmunosupresor rápido6,8.
La inmunoglobulina a dosis de 2 g/kg en 2-5 días es una opción útil cuando los corticoides están contraindicados o cuando está relacionado a anticuerpos NMDA-R. La plasmaféresis (5-10 sesiones) es efectiva también cuando los corticoides están contraindicados o no han sido efectivos. Se ha sugerido que pacientes con encefalitis anti-NMDA-R tratados con corticoides y plasmaféresis tienen mejor respuesta que aquellos tratados solo con corticoides. Otros agentes como rituximab y ciclofosfamida se han usado como segunda línea con buena respuesta6,10,12.
Después de la fase aguda se debe realizar tapering de corticoides. En los casos en que exista recaída o alto riesgo de recurrencia, se puede usar micofenolato mofetilo (MMF) o azatioprina13. También se ha investigado la utilización de metotrexato intratecal, con resultados favorables, pero muy poca experiencia14.
Se han descrito cinco factores de mal pronóstico en la encefalitis autoinmune: status epiléptico refractario, edad de inicio >60 años, subtipo de encefalitis autoinmune mediada por anticuerpos negativa, compromiso infratentorial en RMN cerebral y retraso de inmunoterapia ≥1 mes10. De estas, nuestra paciente reunía dos: subtipo encefalitis autoinmune mediada por anticuerpos negativa y compromiso infratentorial en RMN cerebral, lo que condicionó la persistencia de los síntomas a pesar de completar más de 6 meses de inmunoterapia.
Solo existe, a nuestro entender, un caso reportado en la literatura de encefalitis seronegativa asociada a SSp en una mujer de 31 años con convulsiones focales refractarias, alteraciones cognitivas y RM cerebral con características de encefalitis límbica. Fue tratada con metilprednisolona, plasmaféresis, anticonvulsivantes, y externada con prednisolona oral y MMF 500 mg c/12 h con resolución de los síntomas en el seguimiento a 6 meses7.
La respuesta en nuestro caso fue buena dada la rápida instauración del tratamiento. Sin embargo, se desconoce la duración óptima del tratamiento inmunosupresor, aunque se ha descrito que aproximadamente la mitad de los pacientes puede persistir con síntomas a los 2 años, sin beneficio al continuar la terapia después de un año10.
Conclusiones
La EA seronegativa asociada a SSp es una patología infrecuente que debe sospecharse en pacientes con deterioro cognitivo leve o grave, y que puede estar asociada o no a otros síntomas de actividad de la enfermedad. El tratamiento inmunosupresor, así como el manejo interdisciplinario entre neurólogos, reumatólogos e internistas favorecen el diagnóstico precoz y disminuyen las complicaciones a largo plazo.

